August 31, 2020 report
Mitochondria control cells using their own complete fatty acid synthesis machine
11:e10488, https://doi.org/10.15252/emmm.201910488")
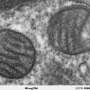
It shouldn't be any secret that mitochondria can make their own fatty acids. The enzymes mitochondria use to do it were discovered decades ago. Unfortunately, only a few individuals among the biologically literate masses have come to appreciate this critical fact about mitochondrial behavior. Perhaps the bigger issue is why mitochondria would go to all the trouble when cells can already make all the fatty acids they need.
Wikipedia itself remains largely in the dark when it comes to mitochondrial fatty acid synthesis. It does contain several exhaustive entries for enzymatic players in the main cellular fatty acid processes, but it is hard pressed even to mention that mitochondria can do it, too. For years, the small cadre of devotees who studied it referred to it as FASII, for fatty acid synthesis type II. This was because it looked just like the pathways of the same name used by bacteria, from which mitochondria are derived. Eukaryotes, on the other hand, employ FAS type I (FASI) in the cytoplasm.
The main difference seems to be that the FASI enzymes have partially merged into large multifunctional conglomerates that carry out whole sequences of reactions together. Presumably, this makes for greater efficiency because the many enzymes and substrates needn't slowly diffuse to find each other within a large cytoplasmic reaction space. In the fullness of time, something curious happened to the field: A trickle of more recent papers began using a new name for FASII as done by mitochondria: It was now mtFAS.
A relevant paper on the topic was just published in the journal eLife by Sara Nowinski, et al. The sole reason for mtFAS has traditionally been to supply an eight-carbon-long fatty chain called octanoic acid. This is, in turn, modified into the lipoic acid cofactor that is used in at least five critical enzymes, all deployed in mitochondria: pyruvate dehydrogenase (PDH), α-ketoglutarate, dehydrogenase (KDH), the glycine cleavage system (GCS), branched chain dehydrogenase (BCD), and α-ketoadipate dehydrogenase (KAD).
It has slowly emerged that mtFAS produces many more longer fatty acids, and is intimately involved with many other important mitochondrial functions like iron-sulfur cluster synthesis and translation by mitoribosomes. What Nowinski's paper nicely demonstrates is that mtFAS has yet another primal role that seems to be independent of making octanoic acid for protein lipoylation—to directly control the electron transport chain itself.
This presents an intriguing chicken and egg situation. It is now appreciated that the single essential function of mitochondria across all organisms that possess them seems not to be electron transport, but rather synthesis of iron-sulfur clusters. This is supported by the fact that when mitochondria have de-evolved in some cells, so to speak, and no longer have the DNA for the critical hydrophobic electron transport proteins, they still have to perform the cluster synthesis. The exception that proves this rule are the amitochondrial creatures that only through incredibly strange twists of evolutionary fate manage to do some rudimentary cluster synthesis in their cytoplasm.
But now, researchers suggest that the saving grace for mtFAS may not be protein lipoylation, or even the cluster synthesis, but rather that it has an accessory electron transport function. The authors got at this problem by engineering cells to express mutated versions of three distinct enzymatic steps in mtFAS; Mcat, Oxsm or Mecr. They also compared cells lacking the terminal step for the lipoylation of enzymes by knocking out Lipt1. These investigations revealed that loss of mtFAS, but not lipoic acid synthesis, impaired the assembly of the electron transport machinery.
The third enzyme mentioned just above, Mecr, is now known to be the problem in a rare orphan disease known as MEPAN syndrome. MEPAN stands for mitochondrial enoyl CoA reductase protein-associated neurodegeneration. The MEPAN Foundation was founded by Danny Miller to find treatments and cures for his sons Carson and Chase. The boys cannot speak or walk, presumably because their basal ganglia circuitry has been severely affected by MEPAN. Although it is not yet understood how or why MEPAN so uniquely targets the basal ganglia, many clues are now emerging.
A recent review on mitochondrial fatty acids and neurodegenerative disorders by Alex Kastaniotis et al. further explores the MEPAN disease ecosystem by looking at what we might call the "metabolic metadata." This is simply a matter of expanding on the metabolic critical points. In other words, looking at next enzymes down the line that depend on the affected product, the ones before it that supply the essential precursors, and also those parallel to it that compete with or complement the pathway at hand. In the case of MEPAN, this metadata includes the related diseases known as PKAN and CoPAN, which stand for Pantothenate kinase-associated neurodegeneration, and for coenzyme A (CoA) synthase protein associated neurodegeneration, respectively.
Pantothenate (vitamin B5) is needed to build CoA, and both PKAN andCoPAN synthesis of CoA is directly disrupted. Deficiencies of pyruvate dehydrogenase (PDH), which is responsible for turning CoA into acetyl-CoA, show a similar phenotype to PKAN and CoPAN. The main finding upon MRI is the accumulation of excessive iron in a region of the basal ganglia known as the globus pallidus. While MEPAN affects the basal ganglia, it is not usually hallmarked by iron accumulation. We might consider MEPAN to be a superset of the PDH disorders in that it also involves four other lipoic-acid-containing enzymes, but there are several key differences.
One paper I recently discussed with Alex on this general topic was that of Lambrecht et. al., which explores several new hypotheses, including the idea that the globus pallidus may be selectively vulnerable to decreased activity of PDH. While all these disorders have been inferred to lead to lack of mitochondrial fatty acids, MEPAN patients differ in that they still have normal levels of a molecule known as (holo-)ACP, rather than severely reduced levels. ACP, or acyl carrier protein, is needed in fatty acid synthesis and also for the iron-sulfur cluster machinery. Alex suggests that a mitochondrial iron overload could occur as a direct response to iron-sulfur cluster deficiency.
Alex notes that the mammalian and yeast mtFAS pathways are structured very similarly and resemble the prokaryotic FASII in that the MCAT, OXSM, and MECR enzymes can all be identified by homology searches in human protein databases using yeast Mct1, Cem1, and Etr1 protein sequences as templates. The former two are also direct orthologs of the corresponding bacterial enzymes, however, the remainder of the members of the mtFAS pathway show an considerable increase in complexity. In particular, the mammalian 3-ketoacyl reductase is composed as a "dimer of dimers" and can be better understood by comparison to microbial ketoreductases.
When I asked Sara Nowinski about homology, she mentioned that there are bacteria with type I FAS, and possibly even bacteria that do both. There are also mammalian enzymes that, while functionally conserved, are not orthologous at all to the bacterial (or even yeast) FASII enzymes, and there are steps in the bacterial pathway that are missing in mtFAS. All this indicates that mitochondria have done something new with fatty acid synthesis that we are only just now beginning to understand.
More information: Sara M Nowinski et al. Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria, eLife (2020). DOI: 10.7554/eLife.58041
Alexander J. Kastaniotis et al. Mitochondrial Fatty Acids and Neurodegenerative Disorders, The Neuroscientist (2020). DOI: 10.1177/1073858420936162
Journal information: eLife
© 2020 Science X Network