First genome of spotted lanternfly built from a single insect

Agricultural Research Service (ARS) scientists, in cooperation with Pacific Biosciences and Penn State University, have published the first genome of the invasive Spotted Lanternfly (SLF) in the journal Gigascience and they did it from a single caught-in-the-wild specimen.
Not only is it the first published genome for this pest, but no closely related species has had its genome sequenced, making the data even more important, according to entomologist Scott M. Geib with the ARS Daniel K Inouye U.S. Pacific Basin Agricultural Research Center.
SLF, a native of China, Bangladesh and Vietnam, was first found in Pennsylvania in 2014 and has now spread to Virginia, Maryland and New York. This invasive pest has a taste for almonds, apples, apricots, grapes, peaches, blueberries and hops as well as hardwoods such as oak, walnut, and poplar. Various estimates put the potential economic damage in the billions of dollars, if the SLF becomes widely established in the United States.
"Having the genome for this pest opens the door to a better understanding of its biology and behavior, and makes coming up with potential control methods much more likely to happen, such as developing a lure for a trap through understanding the insect's olfactory genes, or exploring avenues such as gene editing or RNAi," said Geib.
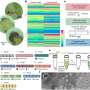
While having the SLF genome is critical for the management and control of this invasive pest, the approach taken to obtain the genetic data is an achievement of remarkable note as well. For the first time, all of the DNA required to generate a whole genome sequence was taken from a single insect picked from a tree in the wild in Reading, Pennsylvania, across the street from the Reading Pagoda on Mt. Penn.
One hurdle for deciphering this species' genome is its relatively large genome size, at about 2.2 billion base pairs. Typically, with previous sequencing systems, many sequencing runs would have been needed to do the complete job, with each run using up the available DNA for the organism being sequenced.
So often to have sufficient DNA for a complete genome sequence, many organisms would need to be pooled, introducing more opportunities for errors to be generated. To avoid such potential for errors, the subjects—especially insects—often have to be raised in colonies and inbred.
"In cooperation with Pacific Biosciences and using their new sequencing platform—the PacBio Sequel II—that produces 10 times the data from a single sequencing run, we were able to generate sufficient coverage from just a single specimen. This allows for a very fast turn-around of data and assemblies as well as lowers cost, in this case under $2,000 in consumable supplies, not including the purchase price of the sequencing instrument of course," explained Geib.
For genome completeness, and since there aren't many related genomes to compare that of the SLF to, the team checked a set of "core genes" that should be present exactly one time in all insects and verified how many of these were found in this genome project. In this case, they found about 97 percent of these single copy core genes, with a very low rate of duplication.
"Sequencing such a large insect genome quickly and showing there is no need to pull the insect into a colony raises the feasibility that we can complete the Ag100Pest Project," Geib said. The ARS Ag100Pest initiative is focused on deciphering the genomes of 100 insect species that are most destructive to crops and livestock and that are projected to have profound bioeconomic impacts to agriculture and the environment. "Now, with this system, doing 100 or even 1,000 genomes is not unrealistic," he added.
The ability to get a complete genome from a small amount of DNA also makes it practical to consider sequencing the genomes of physically tiny insects without having to catch or raise a large number of any one species. That expands the list of insects that may be genetically sequenced.
More information: Sarah B Kingan et al, A high-quality genome assembly from a single, field-collected spotted lanternfly (Lycorma delicatula) using the PacBio Sequel II system, GigaScience (2019). DOI: 10.1093/gigascience/giz122
Journal information: GigaScience
Provided by US Department of Agriculture