Artificial intelligence system designs drugs from scratch
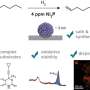
 Training step of the generative Stack-RNN. (B) Generator step of the generative Stack-RNN. During training, the input token is a character in the currently processed SMILES string from the training set. The model outputs the probability vector pΘ(at|st − 1) of the next character given a prefix. Vector of parameters Θ is optimized by cross-entropy loss function minimization. In the generator regime, the input token is a previously generated character. Next, character at is sampled randomly from the distribution pΘ(at| st − 1). (C) General pipeline of RL system for novel compound generation. (D) Scheme of predictive model. This model takes a SMILES string as an input and provides one real number, which is an estimated property value, as an output. Parameters of the model are trained by l2-squared loss function minimization. Credit: Science Advances (2018). DOI: 10.1126/sciadv.aap7885")
An artificial-intelligence approach created at the University of North Carolina at Chapel Hill Eshelman School of Pharmacy can teach itself to design new drug molecules from scratch and has the potential to dramatically accelerate the design of new drug candidates.
The system is called Reinforcement Learning for Structural Evolution, known as ReLeaSE, and is an algorithm and computer program that comprises two neural networks which can be thought of as a teacher and a student. The teacher knows the syntax and linguistic rules behind the vocabulary of chemical structures for about 1.7 million known biologically active molecules. By working with the teacher, the student learns over time and becomes better at proposing molecules that are likely to be useful as new medicines.
Alexander Tropsha, Olexandr Isayev and Mariya Popova, all of the UNC Eshelman School of Pharmacy, are the creators of ReLeaSE. The University has applied for a patent for the technology, and the team published a proof-of-concept study in the journal Science Advances last week.
"If we compare this process to learning a language, then after the student learns the molecular alphabet and the rules of the language, they can create new 'words,' or molecules," said Tropsha. "If the new molecule is realistic and has the desired effect, the teacher approves. If not, the teacher disapproves, forcing the student to avoid bad molecules and create good ones."
ReLeaSE is a powerful innovation to virtual screening, the computational method widely used by the pharmaceutical industry to identify viable drug candidates. Virtual screening allows scientists to evaluate existing large chemical libraries, but the method only works for known chemicals. ReLeASE has the unique ability to create and evaluate new molecules.
"A scientist using virtual screening is like a customer ordering in a restaurant. What can be ordered is usually limited by the menu," said Isayev. "We want to give scientists a grocery store and a personal chef who can create any dish they want."
The team has used ReLeaSE to generate molecules with properties that they specified, such as desired bioactivity and safety profiles. The team used the ReLeaSE method to design molecules with customized physical properties, such as melting point and solubility in water, and to design new compounds with inhibitory activity against an enzyme that is associated with leukemia.
"The ability of the algorithm to design new, and therefore immediately patentable, chemical entities with specific biological activities and optimal safety profiles should be highly attractive to an industry that is constantly searching for new approaches to shorten the time it takes to bring a new drug candidate to clinical trials," said Tropsha.
More information: Mariya Popova et al, Deep reinforcement learning for de novo drug design, Science Advances (2018). DOI: 10.1126/sciadv.aap7885
Journal information: Science Advances
Provided by University of North Carolina at Chapel Hill