May 11, 2018 report
Mitochondria and the art of DNA maintenance

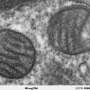
Humans have 46 chromosomes, and each one is capped at either end by repetitive sequences called telomeres. If you ask a biologist if humans have circular DNA, they are likely to say 'no.' That is because eukaryotic cell nuclei have linear chromosomes, while prokaryotes have circular nucleoids and plasmids. However, biologists also know that most of our cells have mitochondria that do, in fact, retain the circular DNA, the chromosome 'M,' which they inherited from their prokaryotic ancestors.
One might then ask: Do mitochondria contain linear DNA? The correct answer to this second, and somewhat sneaky question is, again, affirmative. Nucleoids in mitochondria do need to be circular in order for the machinery that copies their DNA to work. Transcription in mitochondria is directly coupled to replication, and also requires circularized nucleoids. However, linear nucleoids exist in a healthy state of equilibrium with circular nucleoids within the mitochondria network. This provides a way for the cell or tissue to control the abundance of mtDNA directly, and by implication, the state and abundance of mitochondria.
What is the fate of linear mtDNA? Double-strand breaks (DSBs) are continually generated as a byproduct of replication stalling, or from failed DNA repair of damaged and incorrectly replicated nucleotides. Although nucleoids can normally replicate themselves in about 90 minutes, DNA polymerases are hung out to dry when nucleotide stores are insufficient or become improperly balanced. When that happens, things break down; essential factors begin to leave the replication-transcription complex, and proper proofreading becomes a frequent casualty.
Complete double-strand breaks are generally considered to be the worst of the worst when it comes to DNA damage. Loose ends are basically live wires that can readily recombine in dangerous ways to wreak transgenerational havoc. Linear chromosomes in the nucleus use telomeres to guard their loose ends from degradation by ubiquitous nucleases. Replication of these special protective features requires a different process from replicating the rest of the chromosome. While mitochondria are shown to be capable of repairing some DSBs using homologous recombination and DNA ligases, the more common way of dealing with DSBs is to chomp up with exonucleases.
Until recently, it was not understood how linear mtDNA was degraded. Authors of a new paper in Nature have now shown that the same exact machinery responsible for replicating mtDNA also polices it for breaks. The three main proteins involved, the helicase TWNK, the polymerase POLG, and the exonuclease MGME1, were found to bind together into a functional unit. TWNK first acts to unwind the DNA so that the individual strands can be accessed. MGME1, which has a strong bias for operation in the 5' to 3' direction of single-stranded DNA then begins to digest one strand.
The key insight the authors made is that the other strand is simultaneously processed by the native 3' to 5' exonuclease activity built into the primary subunit of the POLG polymerase. Introducing specific mutations to eliminate the exonuclease action resulted in a compromised ability to degrade linear mtDNA. POLG mutations are a frequent cause of mitochondrial disease, especially in mtDNA depletion and deletion disorders. Sizeable databases now exist just to keep track of newly reported POLG variants and all their known effects.

Since the subunits of POLG are encoded in the nucleus (within the mitonuclear genome) it may be possible to correct buggy versions of POLG using CRISPR gene editing techniques. Mutations within the mtDNA itself frequently result in other kinds of mitochondrial disorders. Sometimes, specific combinations of normally benign mtDNA variants can even create disease states. Unfortunately, CRISPR doesn't make much sense for editing the minimal, compact genomes of mitochondria. There are very few candidate sites where CRISPR would even be physically possible in mtDNA. For these kinds of issues, new designer nucleases like TALENs (Transcription Activator-like Effector Nucleases) and ZFNs (Zinc Finger Nucleases) that can cut DNA at specific locations have recently become available.
The beauty of the so-called disposable genome of mitochondria, is that full-blown gene editing isn't necessary to fix disease. All one need do is crack open any offending nucleoids, and then let the endogenous degradation systems do their thing. The main difficulty, here, is figuring out where the bad nucleoids are predominantly located, and how to exclusively target them. It is widely appreciated that different tissues can be preferentially depleted of healthy mitochondria, or selectively accumulate damaged mtDNA. For example, fibroblasts and skeletal muscle are associated with specific bodily pathologies, while the retina and cerebellum with specific insults to nerve function.
Assuming one can replenish mtDNA via direct injection at key access points in the body, it would be possible to improve stocks across the board by alternate cycles of mitochondrial binging and purging. In other words, good mitochondria, in suitably encapsulated from, could be periodically transplanted into blood, CSF, or muscle. Then, after some delay, nuclease therapy could alternately introduce DSBs and a period of subsequent degradation. This would have the effect of diluting away the bad mitochondria to any arbitrarily small concentration, at least in theory. Such a strategy would not even require the nucleases to be perfectly selective in targeting the mutated portions of mtDNA.
Another approach, which we might call "mitochondria immunotherapies," could also be a way to apply more power to the problem of clonally expanding mutant mtDNA. This would operate similarly to the now very successful cancer immunotherapies. In certain disease conditions, such as primary biliary cirrhosis, the immune system mistakenly interprets native mitochondrially localized antigens as foreign enemies. In this case, it is the E2 subunit of the pyruvate dehydrogenase complex found in the mitochondrial matrix that is the culprit. The immune system can also recognize particular de-novo mutated mitochondrial products, as well as prokaryotic features of bare mtDNA. For example, hypomethylated CpG sites or oxidative damaged mtDNA can be highly immunogenic if presented by white blood cells in blood or tissue.
Some additional insights (and warnings) into how double-strand breaks might occur and resolve have been provided by researchers Doug Turnbill and Robert Taylor from the Wellcome Center for Mitochondrial Research. In one particular paper, they proposed that mtDNA deletions are most commonly generated during repair of DNA damage as opposed to replication errors. More specifically, they offer that characteristic deletions are initiated by single-stranded segments of mtDNA that were, in turn, generated by exonucleases attacking double-strand breaks. The free single strands would be able to anneal with microhomology or repeat sequences on other single-stranded mtDNA, and undergo repair to an intact but partially deleted state.
In a later paper, these researchers reported that patients with mitochondrial disease who showed considerable neuronal loss in the substantia nigra all had POLG mutations. Considering the fact that in completely normal and healthy people, about half of all the mtDNA found in neurons of their substantia nigra harbors at least one deletion, developing treatments to fix these predictable insults should be a critical focus of the rapidly developing field of mitochondrial medicine.
More information: Viktoriya Peeva et al. Linear mitochondrial DNA is rapidly degraded by components of the replication machinery, Nature Communications (2018). DOI: 10.1038/s41467-018-04131-w
Journal information: Nature , Nature Communications
© 2018 Phys.org