How are the bacteria in our guts related to each other? New technique provides insight
")
Researchers at the University of California Center for Microbiome Innovation (CMI) have validated a new method for use in microbiome studies that could help detect subtle changes in the composition of a microbial community and provide insight into the evolutionary history of community members. The method is more sensitive than current technologies, and could revolutionize the way microbiome data is analyzed. The findings are published April 17 in mSystems.
When trying to determine the identity of the bacteria that reside in a given environment, such as the human gut, researchers often look for differences in the gene encoding the 16S molecule, which is necessary for bacterial reproduction.
"All bacteria have the 16S gene", said Stefan Janssen, postdoc in the laboratory of CMI director and UC San Diego professor of pediatrics and computer science and engineering Rob Knight. "If they didn't, they couldn't reproduce. We can tell who's who by looking at the nucleotide sequence of this gene."
The sequences are different between bacterial species because the natural process of copying one's DNA is error-prone, and can introduce changes to the genetic material—one of the things that can lead to the rise of new species. Using these sequences, researchers can trace and construct the evolutionary history of the organisms from scratch.
Why is understanding the evolutionary history of organisms important? It's the foundation for innovation, says Janssen.
"We can infer similar functionality for closely related organisms," said Janssen. "This enables us to exploit biology—take what nature has been doing for centuries and maximize its potential for the good of humanity."
But there's a problem with trying to construct the evolutionary history of an organism, or a group of organisms, from scratch using 16S sequences: it's often wrong.
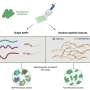
"Scientists sometimes use what's called a 'reference phylogenetic tree'," said Janssen. "Phylogenetic trees are a way for us to visualize the evolutionary history of an organism. They can be useful for determining the complexity of the microbes in a sample (how different they are from each other), and for comparing the microbes in one sample to those in another. Unfortunately, the short sequences generated by 16S sequencing don't contain enough information for us to be able to accurately reference an existing tree."
To solve this problem, Janssen and his team have applied a new algorithm and existing methodology to insert the short fragments of DNA generated by 16S sequencing into reference trees. The technology is more accurate and more sensitive than creating a tree from scratch, which will allow researchers to get more out of the data.
In fact, Janssen says, we might even be able to detect small changes in the gut microbiome that are due to things like dietary changes. "We can easily tell the difference between a skin sample and a stool sample, in terms of microbial community composition. But subtle changes, like who's there when someone changes their diet, may only be detectable with this technology."
The researchers validated the new methodology using bird poop:
Fecal samples were taken from nine different bird species that breed on the Alaska mainland. The birds were in different developmental stages: either hatch year or adult. Using the new methodology, the researchers were able to detect differences in the microbial communities present in the younger birds versus the adult birds—an effect that could not be seen using current methods.
"Our findings have huge implications for pre- and probiotics studies," said Janssen. "The shift in the community may be so small that only this technology could pick up the difference between treatment and control groups."
The paper is titled, "Phylogenetic Placement of Exact Amplicon Sequences Improves Associations with Clinical Information".
More information: Phylogenetic Placement of Exact Amplicon Sequences Improves Associations with Clinical Information, mSystems, DOI: 10.1128/mSystems.00021-18
Provided by University of California - San Diego