Modeling surface chemistry and predicting new materials
The ruddy flakes of a rusted nail are a sure sign that an undesirable chemical reaction has occurred at the surface. Understanding how molecules and atoms behave with each other, especially at surfaces, is central to managing both desirable chemical reactions, such as catalysis, and undesirable reactions, like a nail's corrosion. Yet the field of surface chemistry has been challenged for nearly 100 years to develop predictive theories for these reactions. Now there's progress, thanks to a new approach.
In a presentation at the 64th AVS International Symposium and Exhibition in Tampa, Florida, Oct. 31-Nov. 2, 2017, Alec M. Wodtke and colleagues from the Max Planck Institute for Biophysical Chemistry in Göttingen, Germany, will present what they call a "provisional model" for surface chemistry. In their work they describe how a fruitful interplay between experiment and theory can lead to accurate atomic-scale simulations of simple reactions at metal surfaces.
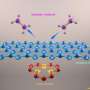
Offering concrete examples, they show that for hydrogen atom interactions with metals—an important approximation in many theories—the Born-Oppenheimer approximation fails for hydrogen atom interactions with metals, but is valid for interactions with graphene. Interestingly, hydrogen interactions on graphene are strongly influenced by the choice of metal substrate upon which the graphene is grown. This makes the study a hot topic because of the potential of graphene in consumer applications, from medical devices to computers.
In another presentation is this surface science session, Arthur L. Utz of Tufts University in Massachusetts and his colleagues will describe the promising results of a collaboration with the Kroes Group of Leiden University, Netherlands, using a new computational approach to predict the reactivity of methane molecules reacting on a clean nickel surface.
Despite significant differences in energy distribution, subsequent calculations yielded chemically accurate predictions of reactivity for thermally excited and vibrational-state-selected molecules, and even for different surface structures, a finding poised to accelerate discovery of materials.
The team's approach allows investigators to predict the fraction of molecules that react on a catalytically active surface with much higher accuracy than has been possible in the past. The results of this research could help accelerate the discovery of new materials.
Provided by AVS: Science and Technology of Materials, Interfaces, and Processing