April 14, 2017 report
New whole genome amplification method reduces biases introduced by other methods

(Phys.org)—A team of researchers working at Harvard University has developed a new whole-genome amplification method that outperforms other methods currently being used. In their paper published in the journal Science, the team describes their technique and how well it performed when used to measure single-nucleotide variations in a human cell after exposure to ultraviolet radiation.
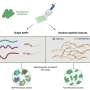
As scientists continue the quest to fully understand the genome, new tools are constantly being developed. One avenue of research involves delving into the differences between individual cells that are almost identical—embryonic cells, for example—each has its own unique genome,even in the same organism. Prior research has led to tools that amplify differences, or variances between cells, which not only helps to better understand how genomes work, but which has practical applications. One such tool that has been developed and used to study and measure genetic variance among individual cells is MALBAC—it is used for in vitro fertilization to screen embryos. But as the researchers also note, it is subject to allele dropout, which limits its ability to call single-nucleotide variations. In this new effort, the researchers have found a way to improve the technique—the improved version is called Linear Amplification via Transposon Insertion (LIANTI), and they claim it has kilobase resolution.
LIANTI works by fragmenting the genetic material in a cell using a transposon the team designed. A transposon is a bit of DNA that is able to alter its position in a genome. It had a 19 base-pair double-stranded transposase binding spot and a single-stranded T7 promoter loop. The transposon carried with it a promoter which was a part of DNA that begins the process of transcription. The promoter was used to amplify the downstream DNA, allowing for the creation of a library that could be sequenced. Initial indications suggested the new technique would outperform other methods.
The researchers tested their technique by exposing human cells to ultraviolet light and then measuring their variances—they report their approach covered 97 percent of the genome, which was significantly better than other methods.
More information: Chongyi Chen et al. Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI), Science (2017). DOI: 10.1126/science.aak9787
Abstract
Single-cell genomics is important for biology and medicine. However, current whole-genome amplification (WGA) methods are limited by low accuracy of copy-number variation (CNV) detection and low amplification fidelity. Here we report an improved single-cell WGA method, Linear Amplification via Transposon Insertion (LIANTI), which outperforms existing methods, enabling micro-CNV detection with kilobase resolution. This allowed direct observation of stochastic firing of DNA replication origins, which differs from cell to cell. We also show that the predominant cytosine-to-thymine mutations observed in single-cell genomics often arise from the artifact of cytosine deamination upon cell lysis. However, identifying single-nucleotide variations (SNVs) can be accomplished by sequencing kindred cells. We determined the spectrum of SNVs in a single human cell after ultraviolet radiation, revealing their nonrandom genome-wide distribution.
Journal information: Science
© 2017 Phys.org