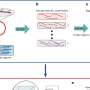
CRISPR-based single-cell genetics platform enables rapid analysis of critical gene networks
Researchers from UC San Francisco and the Broad Institute of MIT and Harvard have developed a new method for performing high-throughput functional screening of complex genetic interactions and resulting phenotypes in single cells, which they have dubbed "Perturb-seq."
The findings could greatly speed scientists' ability to map gene interactions and responses to environmental stimuli to advance understanding of healthy gene networks and how they go awry in the context of disease.
Described in two co-authored papers in the Dec. 15 issue of Cell, one led by Broad Institute researchers and one led by UCSF researchers, the Perturb-seq platform uses single-cell RNA sequencing to measure the effects of many CRISPR-based perturbations on large numbers of cells. The method can be used in many experimental applications, such as exploring the functional impact of genetic risk factors from genomic studies more efficiently than has been previously possible, or looking at genes mutated in cancer cells.
Working collaboratively, the Broad and UCSF teams used Perturb-seq to make new discoveries about, respectively, the immune response in dendritic cells, a cell type that acts as a critical messenger within the immune system, and the unfolded protein response, a cellular stress pathway implicated in a number of neurodegenerative disorders, demonstrating the potential of this platform to yield insight on a variety of biological questions.
"In perturb-Seq, we combined pooled CRISPR screens with the information-rich readout of droplet-based single-cell RNA sequencing, to give us a powerful tool that dramatically increases the scope of what we can learn from functional genomic screens about how circuits are wired inside cells," said Aviv Regev, PhD, senior author of the Broad-led study, a professor of biology at MIT, a core faculty member at the Broad, and a Howard Hughes Medical Institute investigator. "In particular, we can understand how 'the whole is greater than the sum of its parts,' that is, why perturbing two different genes together gives an effect that is different than perturbing each of them alone. This will help us predict better which genes to target in disease as we develop therapies."
"Perturb-seq brings two technologic advances – CRISPR-based perturbations and massively parallel single-cell RNA sequencing – together in a way that we think will greatly speed our ability to understand how different genes that encode for the components of cells are normally wired together, and what goes wrong in human disease," said Jonathan Weissman, PhD, a professor of cellular and molecular pharmacology at UCSF, a Howard Hughes Medical Institute investigator, and senior author of the UCSF-led study.
The design of a robust cell barcoding strategy was also employed to allow the researchers to identify specific perturbations that have been performed on cells within pooled experiments after the fact, while measuring at the same time the effect of those perturbations on the mRNA transcripts in the cell. After an experimental treatment, such as immune stimulation or cellular stress, the cells can be subjected to droplet-based single-cell RNA sequencing to read out both the transcriptional profiles and barcodes' identities. Each cell's transcriptional profile is associated with the one or more genetic perturbations delivered to it by indexing both the cell's mRNA and the barcode identity.
"Functional genomics studies can shed light on the connection between genotype and phenotype, but we'd like to also understand the mechanistic relationships between the two," said Atray Dixit, co-first author of the Broad-led study and a graduate student at MIT. "Looking at the RNA level is a great place to start."
In the Broad-led experiments, researchers used CRISPR/Cas9 nucleases to cut DNA and inactivate genes for transcription factor proteins (TFs) involved in the immune response in dendritic cells, and to inactivate genes for TFs and cell cycle regulators in a cancer cell line. Perturb-seq accurately identified individual gene targets, gene signatures, and cell states affected by the individual gene modifications and explored how these genes interact and depend on one another.
"In the past, functional screens have had to choose between either measuring simple phenotypic changes induced by many perturbations, or taking a rich, high-resolution look at a limited number of perturbations," said Oren Parnas, PhD, co-first author of the Broad-led study, a former postdoctoral researcher at the Broad Institute, and currently a researcher at the School of Medicine of the Hebrew University of Jerusalem. "With Perturb-seq, we can scale up the experiment on both ends."
In the UCSF-led experiments, researchers used CRISPR-based transcriptional interference (CRISPRi) to simultaneously repress up to three genes in a cancer cell line and to investigate the unfolded protein response (UPR), a well-known quality control pathway that senses stress in the endoplasmic reticulum (ER), where many of a cell's key proteins are made. The UPR detects errors in cells' protein-production machinery and ensures that impaired cells self-destruct.
After identifying hundreds of genes whose functions are monitored by the UPR in a genome-wide CRISPRi screen, the UCSF-led team applied Perturb-seq to interrogate a subset of these genes with single-cell resolution, allowing the researchers to systematically reveal the relationships between the genes and dissect the complex ways in which cells respond to ER stress.
"With the unbiased readout we get from single-cell RNA sequencing, we can potentially discover things about biological pathways without a prior hypothesis," said Tom Norman, PhD, co-first author of the UCSF-led study and a postdoctoral researcher in the Weissman lab. "It opens up new possibilities that might not be evident from more targeted studies."
The Perturb-seq method can be used to investigate the functional effects of disease-causing alleles, and to explore on a large scale other targets that could be modified to reverse those effects. The approach is also powerful because it allows the study of several perturbations at once, which can inform the search for combination therapies in cancer, for example.
"By working together to establish the core technology and then pursuing parallel efforts to evaluate Perturb-seq with CRISPR-based methods to inactivate or repress genes in different areas of biology, our teams were able to quickly show that the system is widely applicable," said Britt Adamson, PhD, co-first author of the UCSF-led study and a postdoctoral researcher in the Weissman lab. "The fact that Perturb-seq is not limited to one type of CRISPR-mediated perturbation should be incredibly enabling and allow other research groups to take advantage of the new method."
The researchers say they hope the two Cell studies will serve as robust examples of their new platform, and that as single-cell sequencing technology advances and becomes more powerful and available in the future, Perturb-seq will likely become even easier and faster, enabling increasingly powerful studies that can shed light on genes and their interactions in health and disease.
More information: Atray Dixit et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens, Cell (2016). DOI: 10.1016/j.cell.2016.11.038
Britt Adamson et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response, Cell (2016). DOI: 10.1016/j.cell.2016.11.048
Journal information: Cell
Provided by University of California, San Francisco