November 3, 2016 report
Targeted drug therapy with carrier links to tertiary and heteroaryl amines
 Nature Chemistry (2016) doi:10.1038/nchem.2635")
(Phys.org)—Beyond designing a drug that will attack cancer cells or bacterial cells, drug design also involves figuring out how the drug can safely enter the body and find its target before doing its work. However, many drugs never make it to their desired target either because of solubility issues or inability to access certain parts of the cell. That is where carriers come in. Carriers are like molecular taxis that latch onto a small-molecule drug and transport it to its target location.
These molecular taxis need to connect to the drug in such a way that they safely take it to the location, but the connection must also be reversible so the carrier can eventually release the drug. There are very few types of chemical bonds that can do this and the ones that can are limited in scope.
A group of scientists from Genentech, Inc. have devised a synthetic scheme that is generalizable, protecting-group free, and allows for a carrier linker to attach to tertiary or heteroaryl amines via a quaternary ammonium salt. Additionally, because it is a charged species, it is more water soluble than typical carrier molecules and, therefore, may alleviate the problems associated with aggregation. These linkers can be set up to be cleaved either by proteases in the lysosome or reducing agents in the cytosol. This work appears in Nature Chemistry.
"Enabling the reversible connection of tertiary and heteroaryl amines allows us to consider many new drugs, previously unamenable for targeted therapy," Dr. Thomas Pillow told Phys.org.
The ability to attach a linker to a tertiary amine or a heteroaryl amine is of interest because there are several known anticancer and antibacterial drugs that that have these functional groups. These groups often cannot be converted to a secondary or primary amine because of synthetic complexities or diminished drug potency. These drugs require a carrier molecule in order to be most effective in the body, but most carrier molecules previously could only be connected to primary or secondary amines.
Additionally, many anticancer and antibacterial drugs are hydrophobic. But, the body is a predominantly aqueous environment. This means that, upon entering the body, the hydrophobic molecules tend to aggregate, which can cause problems with bodily systems or protein uptake. Notably, when the Genentech scientists attached a tertiary amine to a linker via a quaternary ammonium salt, it became more hydrophilic suggesting that this might be a good option for countering the aggregation problem.
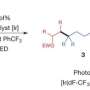
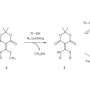
The first step was to determine if a tertiary amine could be reversibly attached to a chemical linker. To test their proposed linker connection, a p-aminobenzyl quarternary ammonium salt, they chose a known dipeptide-cleavable trigger: valine-citrulline (ValCit) which cleaves in the presence of cathepsin B. Incubation of their linker in buffer demonstrated that the quaternary ammonium salts were stable, and exposure to cathepsin B resulted in cleavage at the aniline amide and generation of the tertiary amine as expected.
The next step was to optimize their synthesis to make it simple and generalizable so it can be used for drug screening. In particular, the synthesis could not involve the use of protecting groups and the carrier molecule needed to preferentially react with the tertiary amine on the small molecule drug. After developing the experimental conditions for their reaction, Staben et al. then tested the stability and release of their protease-cleavable linker with a variety of molecules containing tertiary and heteroaryl amines. Good stability and drug release was demonstrated with several of these conjugates.
Thus far, Staben et al. have been looking at the cleavage of peptide bonds. Disulfide bonds are another bond that can be set up as a bioactive trigger that is reduced by intracellular GSH. They tested their system by cleaving the disulfide bond and were able to generate the free tubulysin drug.
In the field of targeted therapies, antibody-drug conjugates have gained ground as a way to target cancer cells or invading bacteria. Prior research by this team demonstrated that an antibody-antibiotic conjugate was able to successfully clear methicillin-resistant S. aureus (MRSA) infections. For the current research, they linked the tertiary amine of a rifamycin analog to their quaternary ammonium salt linker and found that in vitro tests showed a large reduction in the number of intracellular MRSA bacteria, a typically difficult-to-treat bacterial infection.
According to Dr. Pillow, "This novel linker connection expands the universe of drugs for targeted therapy, opening up new possibilities for the treatment of cancer and infectious disease."
More information: Leanna R. Staben et al. Targeted drug delivery through the traceless release of tertiary and heteroaryl amines from antibody–drug conjugates, Nature Chemistry (2016). DOI: 10.1038/nchem.2635
Abstract
The reversible attachment of a small-molecule drug to a carrier for targeted delivery can improve pharmacokinetics and the therapeutic index. Previous studies have reported the delivery of molecules that contain primary and secondary amines via an amide or carbamate bond; however, the ability to employ tertiary-amine-containing bioactive molecules has been elusive. Here we describe a bioreversible linkage based on a quaternary ammonium that can be used to connect a broad array of tertiary and heteroaryl amines to a carrier protein. Using a concise, protecting-group-free synthesis we demonstrate the chemoselective modification of 12 complex molecules that contain a range of reactive functional groups. We also show the utility of this connection with both protease-cleavable and reductively cleavable antibody–drug conjugates that were effective and stable in vitro and in vivo. Studies with a tertiary-amine-containing antibiotic show that the resulting antibody–antibiotic conjugate provided appropriate stability and release characteristics and led to an unexpected improvement in activity over the conjugates previously connected via a carbamate.
Journal information: Nature Chemistry
© 2016 Phys.org