August 9, 2016 report
MIDA boronates react via two different mechanisms
 Nature Chemistry (2016). DOI: 10.1038/nchem.2571")
(Phys.org)—In natural product and pharmaceutical chemistry, one goal is to find a modular reaction to put together complex products, similar to how amino acids combine to form a peptide. Ideally, this modular reaction could couple key building blocks used for many of these processes. N-methylimidodiacetic acid boronic esters, or MIDA boronates, have become the platform of choice for many automated platform small-molecule coupling reactions.
While MIDA boronates are versatile and have many advantages, they have a few limitations that may be overcome with a better understanding of the mechanisms involved in MIDA boronate chemistry. A group of researchers from the University of Edinburgh, Oregon State University, University of Illinois, University of California, Los Angeles, and Liverpool John Moores University have found that MIDA boronates undergo hydrolysis reactions via two different mechanisms: a fast base-mediated mechanism and a slow neutral mechanism. Their work appears in Nature Chemistry.
"The two general mechanisms we have identified for MIDA hydrolysis are intriguingly complex," says Dr. Guy Lloyd-Jones, Forbes Professor of Organic Chemistry at the University of Edinburgh and one of the principle investigators in this research. "Pivotal to our investigation was a virtuous circle of computation and experiment. Perhaps the most surprising observation is that although the intact MIDA moiety is very robust, once one of the two esters has been cleaved (by either mechanism) intramolecular assistance results in rapid cleavage of the second, leading to complete hydrolysis."
One of the advantages of MIDA boronates is that researcher can control the rate of the hydrolysis reaction by changing the reaction conditions. Furthermore, these rates seem to be independent of the particular substituent on the MIDA boronate. This has allowed researchers to control the reaction pathway such that they can build organic molecules in an iterative fashion.
However, MIDA boronates have several limitations. First, cross-coupling reactions that require harsh conditions, such as high temperatures or long reaction times, often result in unwanted hydrolysis reactions. This means that reactions involving saturated carbons or heteroatoms, functional groups that are important to pharmaceutical and natural product chemistry, are not practical because of these unwanted side products.
Secondly, MIDA boronates will also undergo unwanted hydrolysis reactions when assembling the starting material for the cross-coupling reactions.
Thirdly, the deprotection stage can pose a problem depending on the reaction conditions. This requires adjusting the deprotection process for each reaction type, which is not conducive to making a generalizable platform.
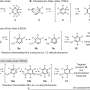
The authors wanted to better understand the MIDA boronate hydrolysis mechanism in hopes of overcoming some of these limitations. They first tested conditions with a model aryl MIDA boronate to discern which ones produced fast or slow reaction rates. They found that there are three distinct hydrolysis reactions: acidic, neutral, and basic. The basic reaction, using NaOH that was slowly added to vigorous stirring, was the fastest. The acidic reaction, using HCl, was only slightly faster than the neutral reaction. The neutral reactions either had K3PO4 or just solvent and water.
Studies with 18O showed that in the basic and acidic reactions one of the esters undergoes a C-O bond cleavage. This is likely due to an OH- mediated reaction. 18O studies were less clear for the slow-release K3PO4 reaction as there was evidence for both mechanisms. The neutral reaction with water showed that the B-O bonds are cleaved during hydrolysis while the esters are not.
Gonzalez, et al. then determined the kinetic rate laws of the three reactions using ultraviolet-visible absorption spectrophotometry at low concentrations using stop-flow techniques. They then analyzed the points of attack on the aryl MIDA boronate using kinetic isotope effect studies. From these studies as well as computational models, they determined two possible mechanisms for MIDA boronate reactions.
The acid and base reaction rates showed little change when the aryl substituent was changed. The acidic mechanism is less sensitive to the substituent than the basic mechanism, but the acid reaction is less efficient. Both indicate a rate limiting ester hydrolysis reaction in which OH- attacks the carbonyl carbon without any involvement from the B-N bond.
The neutral mechanism was tested under several conditions, and while it also displayed a pseudo-first order decay, the rate was less straightforward. Its rate limiting step suggests that the aryl MIDA boronate is attacked by a water cluster. This reaction is more sensitive to the aryl group, likely because the mechanism involves the formation of a boronic anion. Water molecules seem to stretch the B-N bond, and then the bond is cleaved in the rate limiting step.
These studies also showed that the physical conditions in the reaction work up played a role in kinetics. Sodium hydroxide was slowly added to a vigorously stirring solution of reactants. This was necessary to create an emulsified layer. Additionally, the authors point out that by tweaking the aqueous phase in the organic/aqueous reaction matrix will help reduce the incidence of unwanted hydrolysis reactions.
"The mechanistic insight we have gathered should allow the design of a wide range of new MIDA systems with rationally tuned properties," says Dr. Lloyd-Jones. "For example, new ligands that withstand extensive manipulation of their payload yet undergo rapid and quantitative cleavage when required would be highly impactful, and could open the door to a much broader scope of building block-based automated small molecule synthesis."
More information: Jorge A. Gonzalez et al. MIDA boronates are hydrolysed fast and slow by two different mechanisms, Nature Chemistry (2016). DOI: 10.1038/nchem.2571
Abstract
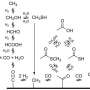
MIDA boronates (N-methylimidodiacetic boronic acid esters) serve as an increasingly general platform for small-molecule construction based on building blocks, largely because of the dramatic and general rate differences with which they are hydrolysed under various basic conditions. Yet the mechanistic underpinnings of these rate differences have remained unclear, which has hindered efforts to address the current limitations of this chemistry. Here we show that there are two distinct mechanisms for this hydrolysis: one is base mediated and the other neutral. The former can proceed more than three orders of magnitude faster than the latter, and involves a rate-limiting attack by a hydroxide at a MIDA carbonyl carbon. The alternative 'neutral' hydrolysis does not require an exogenous acid or base and involves rate-limiting B–N bond cleavage by a small water cluster, (H2O)n. The two mechanisms can operate in parallel, and their relative rates are readily quantified by 18O incorporation. Whether hydrolysis is 'fast' or 'slow' is dictated by the pH, the water activity and the mass-transfer rates between phases. These findings stand to enable, in a rational way, an even more effective and widespread utilization of MIDA boronates in synthesis.
Journal information: Nature Chemistry
© 2016 Phys.org