X-ray laser turns crystal imperfections into better images of important biomolecules
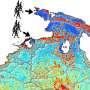
 are displaced from their ideal positions on a regular crystal lattice (tiles). In X-ray crystallography, researchers shine X-rays through crystals to determine the atomic structure of the crystals' units, such as complex biological molecules. A new study of imperfect crystals at SLAC's LCLS X-ray laser has shown that the imperfections can be exploited to obtain much higher-resolution images than with conventional methods. Credit: SLAC National Accelerator Laboratory")
Often the most difficult step in taking atomic-resolution images of biological molecules is getting them to form high-quality crystals needed for X-ray studies of their structure. Now researchers have shown they can get sharp images even with imperfect crystals using the world's brightest X-ray source at the Department of Energy's SLAC National Accelerator Laboratory.
These surprising results could in many cases make the search for better crystals obsolete and fundamentally change the way scientists study the complex biological machinery involved in photosynthesis, catalysis and many other important processes in living things. A better understanding of these processes could drive innovation in a number of areas, from clean energy production to drug development.
"Once the full potential of the new method is understood, it could turn out to be one of the biggest advances since the birth of crystallography," said Mike Dunne, director of the Linac Coherent Light Source (LCLS) X-ray laser, a DOE Office of Science User Facility. The new findings were published today in Nature.
Signals Between Signals
More than 100 years ago, Australian-born British physicist William Lawrence Bragg found a way to use X-rays to probe the interior of crystals, which consist of regular arrays of atoms or molecules. This discovery launched the field of X-ray crystallography, one of the most important techniques for analyzing the structures of materials, chemical processes and biological molecules.
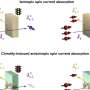
 compared to images obtained from Bragg peaks only (left, mesh). Credit: D. Oberthuer and K. Ayyer/DESY")
For instance, the method has been hugely successful in determining the atomic structures of proteins and shining light on protein function. To date, well over 100,000 protein structures have been determined with X-ray crystallography, which requires many copies of a protein to be incorporated into a single crystal.
When X-rays pass through the crystal, they scatter off the protein molecules and form a pattern on a detector. This diffraction pattern is dominated by bright spots known as Bragg peaks, and researchers use these spots to reconstruct the atomic structure of the molecules.
A perfectly ordered crystal would produce nothing but Bragg peaks. However, disorder limits the number of detectable peaks and therefore the resolution of the molecular image that can be obtained from the peaks alone.
Disorder also produces gently rippling patterns between and beyond the sharp Bragg peaks. While these patterns, known as "continuous diffraction," have been actively studied, they had not been considered capable of producing high-resolution molecular images.
"We've now demonstrated that we can actually use the continuous diffraction of imperfect crystals to obtain better molecular images than with Bragg peaks alone," said Kartik Ayyer, the study's lead author from the Center for Free-Electron Laser Science (CFEL) at the German research center DESY.
The researchers applied their method to crystals of photosystem II - a large protein machine involved in photosynthesis - and found that combining information from Bragg and non-Bragg signals produced higher-resolution images with significantly more structural detail than images obtained with the conventional Bragg-only method.
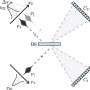
 pass through a crystal (green shapes), they form an intensity pattern on a detector behind the crystal that is dominated by bright spots (dots in the background). Researchers use these so-called Bragg peaks to reconstruct atomic-resolution images of the molecules inside the crystals. In a new study at SLAC's LCLS X-ray laser, researchers used continuous diffraction -- signals found between the spots, which appear here as washed-out, continuous lines in the background -- to improve the resolution of images obtained with the conventional analysis. Credit: SLAC National Accelerator Laboratory")
Crystallography Meets Single-Particle Imaging
The study shows that the continuous diffraction of the photosystem II crystals stems from molecules in the crystal lattice that have shifted out of their ideal positions by as little as the width of a single atom. X-rays scattering off these displaced molecules combine to form the observed continuous pattern rather than Bragg peaks.
"We already know how to analyze these signals," said CFEL scientist Henry Chapman, the principal investigator of the study. "What's special about the continuous diffraction is that it contains significantly more information about the molecular structure than can possibly be measured using Bragg peaks alone. This completely changes our ability to determine the structures of these large, complex biological machines from an almost impossible task to a solvable problem."
One clear benefit of the technique is that it allows researchers, in principle, to reconstruct a biomolecule's atomic structure from scratch, without knowing the locations of some of its atoms or the structure of a similar protein beforehand as a starting point. This would eliminate a major bottleneck.
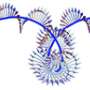
 under X-ray light that contains far more information than the so-called Bragg peaks of a strongly ordered crystal alone (left). The degree of disorder is greatly exaggerated in the crystal on the right. Credit: Eberhard Reimann/DESY")
"The technique is a very elegant marriage between two approaches: X-ray diffraction of crystals and X-ray imaging of single particles," said Ilme Schlichting from the German Max Planck Institute for Medical Research, who was not involved in the study. "It uses the best of both worlds."
The approach could also be a stepping stone toward molecular imaging of single particles, she said. This has been a key goal of modern X-ray science, as it allows measurements of the large number of biological specimens that cannot easily be crystallized. However, single molecules produce notoriously weak diffraction intensities, and it is also challenging to determine individual molecular orientations - a prerequisite for this type of study. Having multiple copies of a molecule in the lattice of imperfect crystals solves both problems.
New Approach with Promising Perspectives
The approach holds the promise to dramatically change the way scientists exploit X-ray lasers for biological studies, and its wider application is currently being assessed. It remains to be seen whether the technique can also be used at synchrotron facilities - X-ray light sources that are less powerful but much more widespread than X-ray lasers.
"Since light from LCLS is so bright, our data could be taken very rapidly and on very small crystals," said LCLS researcher and co-author Sébastien Boutet. "The same experiment at a synchrotron is likely to be more challenging because it would require longer exposures to X-rays, increasing the risk for sample damage, and also require larger crystals that are more likely to show additional unwanted disorder."
Although the researchers have demonstrated their method only on photosystem II, they are confident that it will work for other biomolecules as well. Schlichting agrees. "The kind of disorder used in this research occurs frequently," she said. "It makes the approach an extremely valuable tool."
More information: Macromolecular diffractive imaging using imperfect crystals, Nature, nature.com/articles/doi:10.1038/nature16949
Journal information: Nature
Provided by SLAC National Accelerator Laboratory