July 14, 2015 report
Researchers provide evidence for a non-enzymatic pathway to produce paracaseolide A
 doi:10.1038/nchem.2281")
(Phys.org)—Paracaseolide A is a naturally occurring compound found in the Chinese mangrove plant (Sonneratia paracaseolaris). Among its abilities, it inhibits an important enzyme in the cell cycle progression and affects regulation of the insulin pathway. What is perhaps most fascinating about this compound is its structure. Tao Wang and Thomas R. Hoye from the Department of Chemistry at the University of Minnesota provide compelling evidence that paracaseolide A proceeds via a different reaction mechanism that what has been reported by several groups in the literature. Specifically, they test whether the reaction proceeds via the spontaneous dimerization of a hydroxybutenolide precursor. Their work appears in Nature Chemistry.
Isolation chemists have proposed that paracaseoline A arose through a Diels Alder [4+2] cycloaddition of an alpha-alkenylbutenolide. In all of these studies the researchers completed both the Diels-Alder cycloaddition and dehydration reaction in a single operation under harsh conditions (110oC).
The assumption was that the initial dimerization proceeded via an endo transition state geometry. The harsh conditions that were required to complete this process gave rise to the idea that there is a naturally-occurring enzyme in the mangrove plant to catalyze this dimerization (a Diels-Alderase).
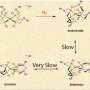
Wang and Hoye report that the dimerization of the alkenylbutenolide precursor can occur through milder conditions than the typical 110oC reported in the literature and that the precursors interact through an exo orientation rather than the reported endo orientation. This means that in order to arrive at the relative configuration of the natural product, one of the carbon atoms undergoes an epimerization. Wang and Hoye provide compelling evidence that this epimerization occurs through a keto-enol tautomerization and subsequent interaction with the solvent.
To prove that the cycloaddition occurs through an exo transition state, Wang and Hoye first investigated an analog to paracaseolide A that has shortened methyl chains rather than the dodecyl alkyl chains. After verifying that their synthetic analog behaved similarly to the naturally-occurring paracaseolide A under the harsh synthetic conditions mentioned in the literature, they tested whether they could obtain product using milder conditions. They were able to isolate the exo Diels-Alder adduct (pre-dehydration intermediate) by reacting the butenolide precursor at 35oC, neat, for six days. The authors verified that it was the Diels-Alder adduct using X-ray crystallography. They also verified that the dimerization step must have proceeded through an exo orientation.
DFT studies showed that the transition state leading to the exo Diels-Alder adduct was lower in energy than that of the endo transition state. Furthermore, tests in which the alkyl truncated analog and the analogous Diels-Alder adduct with the dodecyl alkyl chains in place (which could also be formed under ambient conditions) were mixed showed that the reaction via the exo pathway is not reversible.
Wang and Hoye believe that the exo adduct is preferred over the endo adduct because the geometry of lowest energy transition state is C2-symmetric. The product is formed through a pathway known as a bis-pericyclic process, passing through a transition state in which the two possible modes of cycloaddition [4+2] and [2+4] have merged. This kind of C2-symmetric, bis-pericyclic assembly has been proposed to be operative in the formation of other simpler compounds and to account for their stereochemistry (e.g., dicyclopentadiene), but not for a natural product, here paracaseolide A.
Since one of the four fused rings in the isolated exo adduct is open (a keto acid), Wang and Hoye explored whether reaction through the ring-opened tautomer of the precursor hemiacylal was a possible route for the naturally occurring synthesis. It turns out that this route may be a possibility as it also yields an exo analog for its product.
Finally, optical studies suggest that naturally occurring paracaseolide A is racemic just as the synthetic product is. If this is the case, then the naturally occurring reaction likely did not proceed via an enzymatic pathway. Enzymes typically produce an excess of one enatiomer.
All of these experiments provide evidence that naturally occurring paracaseolide A is likely formed through a different reaction mechanism than what has been presented in the published literature. The synthesis of paracasolide A does not have to proceed via dimerization under harsh conditions producing an endo Diels-Alder adduct, but likely proceeds through a bis-pericyclic [4+2] dimerization. The mild nature of the conditions required for the dimerization as well as the racemic nature of the naturally produced paracaseolide A are consistent with the proposition that this spontaneous dimerization occurs in nature and that dimerization does not require catalysis by a Diels-Alderase enzyme.
More information: "Diels-Alderase-free, bis-pericyclic, [4+2] dimerization in the biosynthesis of (+/-)-peracaseolide A" Tao Wang and Thomas R. Hoye, Nature Chemistry, DOI: 10.1038/nchem.2281
Abstract
The natural product paracaseolide A is a tetracyclic dilactone containing six adjacent stereocentres. Its skeleton occupies a unique structural space among the >200,000 characterized secondary metabolites. Six different research groups have reported a chemical synthesis of this compound, five of which used a thermal, net Diels–Alder [4+2] cycloaddition and dehydration at 110 °C to access the target by dimerization of a simple butenolide precursor. Here, we report that this dimerization proceeds under much milder conditions and with a different stereochemical outcome than previously recognized. This can be rationalized by invoking a bis-pericyclic transition state. Furthermore, we find that spontaneous epimerization, necessary to correct the configuration at one key stereocentre, is viable and that natural paracaseolide A is racemic. Together, these facts point to the absence of enzymatic catalysis (that is, Diels–Alderase activity) in the cycloaddition and strongly suggest that a non-enzyme-mediated dimerization is the actual event by which paracaseolide A is produced in nature.
Journal information: Nature Chemistry
© 2015 Phys.org