Best of both worlds: Hybrid approach sheds light on crystal structure solution
Understanding the arrangement of atoms in a solid—one of solids' fundamental properties—is vital to advanced materials research. For decades, two camps of researchers have been working to develop methods to understand these so-called crystal structures. "Solution" methods, championed by experimental researchers, draw on data from diffraction experiments, while "prediction" methods of computational materials scientists bypass experimental data altogether.
While progress has been made, computational scientists still cannot make crystal structure predictions routinely. Now, drawing on both prediction and solution methods, Northwestern University researchers have developed a new code to solve crystal structures automatically and in cases where traditional experimental methods struggle.
Key to the research was integrating evidence about solids' symmetry—the symmetrical arrangement of atoms within the crystal structure—into a promising computational model.
"We took the best of both worlds," said Chris Wolverton, professor of materials science and engineering at Northwestern's McCormick School of Engineering and expert in computational materials science. "Computational materials scientists had developed a great optimization algorithm, but it failed to take into account some important facts gathered by experimentalists. By simply integrating that information into the algorithm, we can have a much fuller understanding of crystal structures."
The resulting algorithm could allow researchers to understand the structures of new compounds for applications ranging from hydrogen storage to lithium-ion batteries.
A paper describing the research, "A Hybrid Computational-Experimental Approach for Automated Crystal Structure Solution," was published November 25 in the journal Nature Materials.
While both computational and experimental researchers have made strides in determining the crystal structure of materials, their efforts have some limitations. Diffraction experiments are labor-intensive and have high potential for human error, while most existing computational approaches neglect potentially valuable experimental input.
When computational and experimental research is combined, however, those limitations can be overcome, the researchers found.
In their research, the Northwestern authors seized onto an important fact: that while the precise atomic arrangements for a given solid may be unknown, experiments have revealed the symmetries present in tens of thousands of known compounds. This database of information is useful in solving the structures of new compounds.
The researchers were able to revise a useful model—known as the genetic algorithm, which mimics the process of biological evolution—to take those data into account.
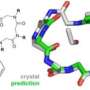
In the paper, the researchers used this technique to analyze the atomic structure of four technologically relevant solids whose crystal structure has been debated by scholars—magnesium imide, ammonia borane, lithium peroxide, and high-pressure silane—and demonstrated how their method would solve their atomic structures.
Bryce Meredig (PhD '12) was the paper's lead author.
Journal information: Nature Materials
Provided by Northwestern University