Predicting protein binding sites on DNA

In silico prediction of protein folding has the potential to reveal the specificity of a given protein sequence for DNA. Such methods are particularly promising as they could open the road to the rational design of novel regulatory molecules.
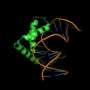
Studying the specificity of protein-DNA interactions has important ramifications for the analysis and prediction of the gene regulatory networks that govern several crucial biological processes. Approaches that rely on the structure of proteins are becoming increasingly promising since they can predict previously undetected binding sites.
The European 'Inferring DNA binding specificities through in silico folding of natively unstructured protein regions' (PROTDNABINDSPEC) project aimed at expanding our current knowledge of protein-DNA binding modes. The plan was to use structural bioinformatics methods in order to predict the structure and specificity of DNA-binding proteins, focusing on natively unfolded protein regions. Such regions consist of flexible segments that do not assume a fixed conformation in the native state, but fold upon binding.
To achieve this, scientists modelled the interaction energy between different amino acids and nucleotides. These statistical potentials were integrated into Fragfold, one of the first fragment-based platforms for molecule fold prediction. The generated complexes would be used to predict DNA binding sites in genomes.
The results of the PROTDNABINDSPEC project are expected to improve our understanding of macromolecular interactions, enabling the annotation of genomic sequences and assisting future research in systems biology.
Provided by CORDIS