Making sense of molecular fragments
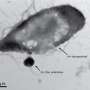
, expressed in different plant tissues and conditions, the ARTADE2 computational algorithm produces more accurate representations of RNA molecule reconstructions (bottom) than the pre-processed transcriptional expression level indications shown in red (top). Unclear representations of the genes (top left), for example, are visible after applying ARTADE2 (bottom left). Credit: 2012 Tetsuro Toyoda, RIKEN Yokohama Institute")
(Phys.org) -- Data from high-throughput next generation sequencers (NGS) and genome tiling arrays have greatly enhanced scientists’ ability to recreate RNA molecular structures, which is vital to disease and biotechnology research. However, high levels of noise and bias in some processes lead to uneven gene-expression values for segments belonging to the same molecule. Reconstructing the complete, or ‘full-length’, information of molecules as they occur in cells is therefore difficult.
To improve accuracy by reducing noise and bias, Tetsuro Toyoda, Shuji Kawaguchi, and Kei Iida at the RIKEN Bioinformatics And Systems Engineering division (BASE) in Yokohama, together with scientists from the RIKEN Plant Science Center, have developed a statistical algorithm for reconstructing full-length information of RNA molecules using output from tiling arrays and NGS. They implemented this algorithm in a computer program called ‘ARabidopsis Tiling-Array-based Detection of Exons’ (ARTADE).
The genome encoded in an organism’s DNA holds the blueprint for building and maintaining cells. For this building and maintenance to work, the DNA blueprint is copied, or ‘transcribed’, by molecules of RNA ‘transcripts’. RNA molecules use this code to create proteins or act themselves as functional molecules and regulate cell activities. Transcriptome is the name given to all the transcripts present at any one time in a cell. Transcriptomes hold vital information about living organisms, including how different protein genes are switched on and off in response to different environmental stresses.
Toyoda and his team further developed ARTADE, ARTADE2, so they could rebuild a virtual representation of the transcriptomes comprising RNA molecules. “Understanding transcriptomes is essential for research on molecular mechanisms of diseases and development of biotechnology with plant species,” Toyoda explains. “Both genome tiling arrays and NGS have output problems with uneven expression values from fragmentation and noise and bias from machinery. This makes it difficult to form a perfect reconstruction.”
ARTADE2 uses a new ‘positional correlation analysis’ developed by Kawaguchi and Iida so that it can analyze any species and be used for NGS output (Fig. 1). This process identifies areas where the transcriptional activities among multiple cellular conditions—such as differences in tissues, developmental stage, or environment—are highly correlated. Positional correlation removes output problems, providing a better representation of the original molecules.
The team has now begun developing a database using information from the rebuilt transcriptomes. This new technology and database will lead to deeper understanding of molecular structures and their alteration according to environmental stresses and disease, furthering understanding of the relationship between genome sequences and cell activities.
More information: Kawaguchi, S., et al. Positional correlation analysis improves reconstruction of full-length transcripts and alternative isoforms from noisy array signals or short reads. Bioinformatics 28, 929–937 (2012). doi: 10.1093/bioinformatics/bts065
Journal information: Bioinformatics
Provided by RIKEN









.jpg)