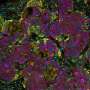
Evolution reveals missing link between DNA and protein shape
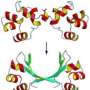
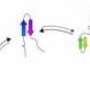
 that seemed to change in lockstep in the evolutionary record. These co-varying pairs indicated points on protein (middle) likely to be in contact after folding, giving researchers enough clues to create a computational model of the protein's three-dimensional structure (right). Credit: Terry Helms/Memorial Sloan-Kettering Cancer Center")
Fifty years after the pioneering discovery that a protein's three-dimensional structure is determined solely by the sequence of its amino acids, an international team of researchers has taken a major step toward fulfilling the tantalizing promise: predicting the structure of a protein from its DNA alone.
The team at Harvard Medical School (HMS), Politecnico di Torino / Human Genetics Foundation Torino (HuGeF) and Memorial Sloan-Kettering Cancer Center in New York (MSKCC) has reported substantial progress toward solving a classical problem of molecular biology: the computational protein folding problem.
The results will be published Dec. 7 in the journal PLoS ONE.
In molecular biology and biomedical engineering, knowing the shape of protein molecules is key to understanding how they perform the work of life, the mechanisms of disease and drug design. Normally the shape of protein molecules is determined by expensive and complicated experiments, and for most proteins these experiments have not yet been done. Computing the shape from genetic information alone is possible in principle. But despite limited success for some smaller proteins, this challenge has remained essentially unsolved. The difficulty lies in the enormous complexity of the search space, an astronomically large number of possible shapes. Without any shortcuts, it would take a supercomputer many years to explore all possible shapes of even a small protein.
"Experimental structure determination has a hard time keeping up with the explosion in genetic sequence information," said Debora Marks, a mathematical biologist in the Department of Systems Biology at HMS, who worked closely with Lucy Colwell, a mathematician, who recently moved from Harvard to Cambridge University. They collaborated with physicists Riccardo Zecchina and Andrea Pagnani in Torino in a team effort initiated by Marks and computational biologist Chris Sander of the Computational Biology Program at MSKCC, who had earlier attempted a similar solution to the problem, when substantially fewer sequences were available.
"Collaboration was key," Sander said. "As with many important discoveries in science, no one could provide the answer in isolation."
The international team tested a bold premise: That evolution can provide a roadmap to how the protein folds. Their approach combined three key elements: evolutionary information accumulated for many millions of years; data from high-throughput genetic sequencing; and a key method from statistical physics, co-developed in the Torino group with Martin Weigt, who recently moved to the University of Paris.
Using the accumulated evolutionary information in the form of the sequences of thousands of proteins, grouped in protein families that are likely to have similar shapes, the team found a way to solve the problem: an algorithm to infer which parts of a protein interact to determine its shape. They used a principle from statistical physics called "maximum entropy" in a method that extracts information about microscopic interactions from measurement of system properties.
"The protein folding problem has been a huge combinatorial challenge for decades," said Zecchina, "but our statistical methods turned out to be surprisingly effective in extracting essential information from the evolutionary record."
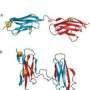
With these internal protein interactions in hand, widely used molecular simulation software developed by Axel Brunger at Stanford University generated the atomic details of the protein shape. The team was for the first time able to compute remarkably accurate shapes from sequence information alone for a test set of 15 diverse proteins, with no protein size limit in sight, with unprecedented accuracy.
"Alone, none of the individual pieces are completely novel, but apparently nobody had put all of them together to predict 3D protein structure," Colwell said.
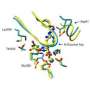
To test their method, the researchers initially focused on the Ras family of signaling proteins, which has been extensively studied because of its known link to cancer. The structure of several Ras-type proteins has already been solved experimentally, but the proteins in the family are larger--with about 160 amino acid residues--than any proteins modeled computationally from sequence alone.
"When we saw the first computationally folded Ras protein, we nearly went through the roof," Marks said. To the researchers' amazement, their model folded within about 3.5 angstroms of the known structure with all the structural elements in the right place. And there is no reason, the authors say, that the method couldn't work with even larger proteins.
The researchers caution that there are other limits, however: Experimental structures, when available, generally are more accurate in atomic detail. And, the method works only when researchers have genetic data for large protein families. But advances in DNA sequencing have yielded a torrent of such data that is forecast to continue growing exponentially in the foreseeable future.
The next step, the researchers say, is to predict the structures of unsolved proteins currently being investigated by structural biologists, before exploring the large uncharted territory of currently unknown protein structures.
"Synergy between computational prediction and experimental determination of structures is likely to yield increasingly valuable insight into the large universe of protein shapes that crucially determine their function and evolutionary dynamics," Sander said.
More information: "Protein 3D structure computed from evolutionary sequence variation," Marks et al. PLoS ONE, December 6, 2011
Journal information: PLoS ONE
Provided by Harvard Medical School