Safeguarding genome integrity through extraordinary DNA repair
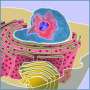
 accounts for a third of the chromatin in both humans and fruit flies. Some heterochromatin forms the telomeres that cap the ends of the chromatids, and much is concentrated near the centromere, where sister chromatids are joined. Accurate repair of double-strand breaks in heterochromatin is challenging, because most of its DNA consists of short, repeated sequences.")
(PhysOrg.com) -- DNA is under constant attack, from internal factors like free radicals and external ones like ionizing radiation. About 10 double-strand breaks – the kind that snap both backbones of the double helix – occur every time a human cell divides. To prevent not only gene mutations but broken chromosomes and chromosomal abnormalities known to cause cancer, infertility, and other diseases in humans, prompt, precise DNA repair is essential.
Scientists at the U.S. Department of Energy’s Lawrence Berkeley National Laboratory (Berkeley Lab), working with cell lines of the fruit fly Drosophila melanogaster, have discovered an unsuspected and dramatic process by which double-strand breaks in heterochromatin – one of the two major kinds of chromatin that make up chromosomes, which accounts for a third of the chromatin in both humans and fruit flies – are repaired in a series of steps. The repair starts where the break occurs, but stalls until the repair site physically moves away from the original heterochromatin region, before continuing to completion.
Unlike euchromatin, where most of an organism’s genes reside and where most DNA consists of long, unrepetitive sequences of base pairs, DNA in heterochromatin consists mostly of short repeated sequences that don’t code for proteins; indeed, heterochromatin was long regarded as containing mostly “junk” DNA.
Heterochromatin is now known to be anything but junk, playing a crucial role in organizing chromosomes and maintaining their integrity during cell division. It is concentrated near centromeres, where chromatids are in closest contact, which are required to transmit chromosomes from one generation to the next. Maintaining heterochromatin structure is necessary to the normal growth and functions of cells and organisms.
“Heterochromatin poses more of a problem for DNA repair than euchromatin,” says Gary Karpen, whose group in Berkeley Lab’s Life Sciences Division discovered the new repair mechanism. “It has lots of short sequences – many of them only about five base-pairs each – which are repeated millions of times.”
“Repair of simple repeated sequences is particularly challenging,” says Irene Chiolo, first author of the group’s paper reporting the results in the journal Cell. “They can promote chromosome aberrations, with severe consequences for the genome stability of dividing cells” – abnormalities that are a hallmark of cancer cells and cause birth defects.
Finding the right path
With the stakes so high, how can cells insure fast, accurate repair of double-strand breaks? Two main repair pathways are available. One method, nonhomologous end-joining, simply cleans up the ends of the broken strands and glues them back together regardless of sequence. This might seem a good choice for heterochromatin: it almost always creates small deletions or mutations, but these are in repetitive, noncoding sequences and do not affect genes.
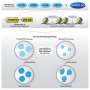
Far more accurate but more complex is homologous recombination, a mechanism involving many steps where something could go wrong. Upon detecting a double-strand break in DNA, several proteins rush to the damaged area. The protein machinery trims back the ends of the broken strands (called ‘resection’)to produce single-strand regions recognized by other proteins, including one called ATRIP.
Another protein, Rad51, is recruited to form filaments on the single-stranded DNA. Rad51 and its associated proteins search for a complementary sequence of DNA in a neighboring chromatid or homologous chromosome. They invade and open that DNA to form a “D-loop” – like untwisting a rope to open and expose its individual strands. Using the exposed complementary sequence as a template, proteins rebuild the broken DNA into a copy of the sequence that was originally damaged; in this way the broken double strand is remade with its damaged section accurately reproduced.
It’s an ideal method for repairing breaks in gene-rich euchromatin. In repetitive heterochromatin, however, danger arises because completely different chromosomes lying close to the site of the break may have great lengths of repeated short sequences that look identical to the region around the break itself. What starts as a repair process may end up splicing different chromosomes together, a common abnormality in cancer cells.
 first trim back the ends of the broken strands (‘resection’), which are recognized by other proteins including ATRIP. Still more proteins, including Rad51, are recruited to form filaments that invade a neighboring chromatid or homologous chromosome having the complementary sequence. Both filaments use these templates (only one filament is shown working here) to accurately reproduce the damaged double strand.")
For heterochromatin to employ such a potentially risky repair process seemed counterintuitive. In earlier experiments looking for key signs of repair in mouse heterochromatin after irradiation, classic markers of double-strand break repair by either nonhomologous end-joining or homologous recombination were both absent. In fact it seemed possible that, somehow, such breaks didn’t occur in heterochromatin.
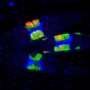
“There were no signs of repair half an hour after the cells were exposed to ionizing radiation,” says Karpen. “But our group looked at Drosophila cells just 10 minutes after radiation exposure. Now the early signs of homologous recombination were clearly evident.”
After half an hour, however, these signs too – signals from modified histones, the component proteins that form the “spools” around which the DNA “thread” is wound in chromatin, as well as signals from ATRIP recruitment – were missing from the heterochromatin domain. What had become of the repair process?
Now you see it, now you don’t
In a series of experiments, Karpen and Chiolo and their colleagues found that in heterochromatin the early stages of homologous recombination – resection and ATRIP loading – appear within three minutes after the damage occurs. The next steps in homologous recombination seemed blocked from entering the heterochromatin at this stage. These steps – the activity of the Rad51 proteins in preparing invasive filaments of single-strand DNA – are the most dangerous, where mistaken recombination could easily occur.
By 30 minutes after radiation damage was inflicted, the entire domain had swollen and began sending out expanding and contracting ‘fingers’ of chromatin. Now, after the relocation of the damaged DNA to outside the heterochromatin, the Rad51 proteins did appear; the researchers found them moving with the ends of the heterochromatin ‘fingers’. After an hour the whole domain partially contracted again, indicating that the broken DNA had moved to the periphery of the heterochromatin domain in order to load Rad51 protein and complete the repair process.
“There are a lot of moving parts here,” says Karpen. “It opens new ways of thinking about DNA repair and investigating the process.”
It’s common to picture chromosomes as rather floppy tubes of stuff that are tightly cinched in their middles to form X-shaped figures, but in fact this is a condensed state that occurs only briefly during mitosis, when cells divide. Most of the time chromosomes aren’t condensed – instead they exist as somewhat diffuse clouds of DNA.
“In the last 20 years researchers have found that the DNA for each chromosome occupies a separate domain in the nucleus, even when chromosomes are decondensed,” says Chiolo. “From these ‘chromosomal territories’ the DNA moves to accomplish certain functions, for example gene transcription, by going to where the proteins are. We now observe that similar movements occur even during DNA repair.”
Says Karpen, “The process we discovered is an extreme version of this dynamism, where the DNA repair process starts in one domain, then the damaged DNA goes elsewhere to complete repair. It would seem that starting repair in one place, then moving elsewhere is risky, and could result in unrepaired damage, which is just as dangerous to the cell as abnormal recombination with a different chromosome.”
Says Chiolo, “Stability is the key. The presence of resected DNA ends is extremely dangerous in heterochromatin only if Rad51 is loaded onto broken DNA.” Sure enough, the researchers discovered that a protein complex called Smc5/6 blocks Rad51 recruitment until the damaged DNA is moved outside the heterochromatin.
Chiolo says, “This mechanism is crucial for safeguarding the genome by blocking aberrant recombination between different chromosomes, and promoting safe repair from a sister chromatin, or homolog, after the double-strand break has relocated outside the heterochromatin domain”.
Homologous recombination is a complex mechanism with multiple steps, but also with many points of regulation to insure accurate recombination at every stage. This could be why this method has been favored during evolution. The machinery that relocalizes the damaged DNA before loading Rad51 might have evolved because the consequences of not having it would be terrible.
Karpen and Chiolo and their colleagues are now at work on the next steps in the research, investigating the many unanswered questions about how this surprisingly dynamic mechanism of DNA repair works and what happens when it fails. Perhaps most important is learning whether the unexpectedly sophisticated approach to homologous recombination in Drosophila heterochromatin is conserved in other organisms, including humans.
“Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair,” by Irene Chiolo, Aki Minoda, Serafin U. Colmenares, Aris Polyzos, Sylvain V. Costes, and Gary H. Karpen, appears in the March 4, 2011 issue of Cell and is available online to subscribers.
Provided by Lawrence Berkeley National Laboratory