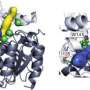
Enzyme design with remote effects
 are drawn together through the substitution of two amino acids (left, red dots) with the result that the remote binding pocket enlarges due to an induced allosteric effect (right, white). Credit: Image: MPI for Coal Research")
Engineers are unlikely to tinker with the cooling system if they want to increase the size of an engine. Yet chemists at the Max Planck Institute for Coal Research have adopted an approach similar to this in their efforts to optimise an enzyme for practical applications. They substituted two amino acids at a site relatively distal to the biocatalyst's binding pocket, the location where the chemical reaction takes place.
This modification alters the overall structure of the enzyme in such a way that it can now convert a larger number of different chemical compounds. In addition, it produces preferentially one of two possible enantiomers which arise as what is known as a racemic mixture during traditional synthesis. Although they are chemically identical, the enantiomers differ in terms of their structure, like a right and left hand. Only one of the two versions is usually suitable for use as a medically active agent.
Thanks to their research, the Max Planck researchers have succeed in pointing the way to a new approach that will enable the conversion of enzymes tailored to the needs of the chemical and pharmaceutical industries. (Proceedings of the National Academy of Science early edition, January 27, 2010)
Enzymes work extremely reliably, however they are highly selective. They transform molecules that arise in nature into the precise form required by a cell or organism. However, they often only convert a single chemical compound - like a garage that would specialise only in the repair of a particular model of vehicle. This is important in biology, as chemical aberrations can paralyse the machinery of the biomolecules. The chemical and pharmaceutical industries need catalysts, for example enzymes, which also process molecules that do not occur in nature. The enzymes involved here are often enantiomers, two deceptively similar compounds that differ like a right and left hand. Chemists are now facing the challenge of finding catalysts that only allow one enantiomer form to arise during a conversion process, that is, catalysts which have an enantioselective effect. Enzymes can sometimes be misappropriated as catalysts to obtain intermediate compounds through enantioselective synthesis for the production of drugs. This process often fails, however, as the enzyme is incapable of recognising an enantiomer form: only one of two enantiomers should arise in the chemical reaction as, in most cases, only one displays the desired biological activity.
Researchers at the Max Planck Institute for Coal Research have now constructed an enzyme that converts numerous molecules enantioselectively with the help of directed evolution. "What we are doing here is bringing about evolution in a test tube," says Manfred T. Reetz, Director of the Max Planck Institute in Mülheim and who heads the current research activities. Hence, the chemists generate enzyme variants by gradually altering their structure through random mutations. Prior to this, they had already structurally modified or designed enzymes enantioselectively by causing random mutations in the active centre where the reaction occurs. They have now developed a fundamentally different strategy, however: the enzyme is altered at a site that is not involved in the actual reaction. "We have adopted a new directed evolution approach here and combined mutagenesis with rational design," says Manfred Reetz.
Reetz and his colleagues started with a natural Baeyer-Villiger monooxygenase. This enzyme oxidises a substrate molecule, a ketone to be more precise, inserting an oxygen atom at a precisely defined site. The product of this process is a lactone. Enantioselectively produced lactones are highly-prized intermediate compounds in the chemical and pharmaceutical industries.
The natural form of an enzyme often works completely non-selectively, that is, it cannot differentiate between enantiomers as both forms are converted with equal speed in the binding pocket. The situation is even more critical when the synthetic molecule is not even converted, as it does not fit in the binding pocket. "To enlarge this binding pocket for synthetic molecules we began by looking for a site in a structural model of the enzyme, in which changes reshape the binding pocket even though it is located at a relatively significant distance from the binding pocket," explains Juan Pablo Acevedo, who, together with Sheng Wu, was closely involved in the reshaping of the enzyme.
They suspected that the site at which amino acid substitution would display the desired effect is located at positions 93 and 94 in the amino acid chain. Based on theoretical considerations, the chemists came to the conclusion that suitable mutations should prompt structural reorganisation there with the result that two domains, that is two sub-units of the enzyme, move together: "Our thinking was that the binding pockets would have to enlarge if we pulled these two domains closer together," relates Juan-Pablo Acevedo. The researchers achieved this by simultaneously substituting the amino acids at the two positions with new agents by means of saturation random mutagenesis. The binding pocket actually enlarged when they inserted asparagine in the place of glutamine in position 93 and aspartic acid in the place of proline in position 94.
The enzyme then converted an entire series of different substances with high activity, and unusually high enantioselectivities were observed. The scientists succeeded in mastering something of a tightrope walk here: while a bigger binding pocket opens up the enzyme for a wider range of substrate molecules, it usually also reduces its stereocontrol over the reaction - a problem did not arise here, however.
What is far more important for the researchers is that their approach basically works: that is, they can reshape the reaction centre by causing a structural change through remote mutagenesis. Biochemists are already familiar with so-called allosteric effects, that is, reorganisation in many enzymes which are accompanied by structural change in the relevant binding pocket. However, these effects are generated by small molecules that dock onto an enzyme far away from the binding pocket. "Using genetic technology methods, we have generated a targeted allosteric effect in an enzyme that is not actually allosteric," says Juan Pablo Acevedo. The Mühlheim-based researchers had already occasionally observed remote mutations in the course of the directed evolution of enantioselective enzymes; however, it had not been hitherto possible to attribute them to allosteric effects.
The altered enzymes are formed using re-programmed bacteria. The researchers infiltrate genetic maps for variants of a natural enzyme into the single-celled organisms. In these maps, they substitute the proposed amino acids at one or more sites. Instead of testing all 20 amino acids from which proteins are constructed, however, they only use twelve. "This cocktail is highly representative of a mixture of structurally and electronically different amino acids," explains Manfred Reetz. Theoretically, the monooxygenase, which he and his colleagues had adopted as the model enzyme, has 144 variants with different amino acid combinations at positions 93 and 94. To actually obtain all of these variants, the scientists must breed approximately 400 colonies from the mixture of reprogrammed bacteria, isolate the enzymes from it and test it for their characteristics.
In this respect, four hundred variants is a small number. If the researchers do not carefully select the positions at which the changes promise to yield the desired effect, and instead apply "blind" random mutagenesis to the entire enzyme, the approach often adopted in directed evolution, they would probably have to test at least several hundred thousand variants - too many for the method to be practicable for the chemical and biotechnology industries. "By combining rational design and random mutagenesis, we can considerably accelerate the development of enantioselective and thermostable enzymes," says Manfred Reetz. This is how he and his colleagues will also proceed in their future work on the conversion of enzymes. They will then combine the changes away from the binding pocket with reorganisation in the reaction centre itself. "We hope that this will enable us to achieve even greater enzyme optimisation," says Manfred Reetz.
More information: Sheng Wu, Juan Pablo Acevedo, und Manfred T. Reetz, Induced allostery in the directed evolution of an enantioselective Baeyer-Villiger monooxygenase, Proceedings of the National Academy of Sciences, in press; doi:10.1073/pnas.0911656107
Provided by Max-Planck-Gesellschaft