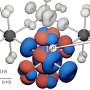
Aging cells lose their grip on DNA rogues

(Phys.org)—Transposable elements are mobile strands of DNA that insert themselves into chromosomes with mostly harmful consequences. Cells try to keep them locked down, but in a new study, Brown University researchers report that aging cells lose their ability to maintain this control. The result may be a further decline in the health of senescent cells and of the aging bodies they compose.
Even in our DNA there is no refuge from rogues that prey on the elderly. Parasitic strands of genetic material called transposable elements—transposons—lurk in our chromosomes, poised to wreak genomic havoc. Cells have evolved ways to defend themselves, but in a new study, Brown University researchers describe how cells lose this ability as they age, possibly resulting in a decline in their function and health.
Barbara McClintock, awarded the Nobel Prize in 1983, made the original discovery of transposons in maize. Since then scientists have found cases in which the chaos they bring can have long-term benefits by increasing genetic diversity in organisms, but in most cases the chaos degrades cell function, such as by disrupting useful genes.
"The cell really is trying to keep these things quiet and keep these things repressed in its genome," said John Sedivy, professor of medical science in the Department of Molecular Biology, Cell Biology, and Biochemistry and senior author of the new study published online in the journal Aging Cell. "We seem to be barely winning this high-stakes warfare, given that these molecular parasites make up over 40 percent of our genomes."
Cells try to clamp down on transposons by winding and packing transposon-rich regions of the genome around little balls of protein called nucleosomes. This confining arrangement is called heterochromatin, and the DNA that is trapped in such a tight heterochromatin prison cannot be transcribed and expressed.
What the research revealed, however, is that carefully maintaining a heterochromatin prison system is a younger cell's game.
"It's very clear that chromatin changes profoundly with aging," Sedivy said.
What Sedivy, lead author Marco De Cecco, and their co-authors measured in several experiments was that young and spry cells distinctly maintain open "euchromatin" formations in regions where essential genes are located and closed "heterochromatin" formations around areas with active transposable elements and few desirable genes.
The distinction appeared to become worn in aging, or senescent, cells. In the observations, the chromatin that once was open tended to become more closed and the chromatin that was once closed, tended to become more open.
Working with computational biologist and Nicola Neretti, assistant professor of biology, Sedivy and De Cecco conducted a genome-wide analysis of these differences. The team extracted and then sequenced DNA from young and senescent human fibroblast cells using a technique called FAIRE. Essentially FAIRE uses chemicals such as formaldehyde to separate out DNA that is loosely packed in euchromatin from DNA that is more tightly wound up in heterochromatin.
Then the scientists compared the DNA that was coming from open or closed chromatin formations in the young and senescent cells.
"Given that our genomes contain well over a million copies of transposable elements and that they are very similar to one another, tracking all this mayhem is no easy matter," Neretti said. "Computationally speaking, it's a nightmare."
But Sedivy said results were well worth the effort. In their study not only did they find that the chromatin lockdown was breaking down, but also that the newly freed transposons were taking full advantage.
"I was really surprised to see that first of all these transposable elements start to get expressed and that they actually start moving around [to other regions in the genome]," Sedivy said. "That's really an amazing thing."
How bad and how to stop it?
What's not clear from the study is the relevance of the damage that the cells suffer from the transposable element jailbreak and resulting genetic crime spree. That depends on the timing, which Sedivy's team measured only in approximate terms.
"Is the transposition really bad for the organism or is it something that happens so late that by that point the organism has already accumulated so much age-associated damage?" he asked. "Then maybe this extra insult of transposition is not going to make a lot of difference."
The question matters, Sedivy says, because drugs might be able to suppress transposons in aging cells. Virtually all of the transposons of concern in mammals are so-called "retrotransposons" because they use RNA and an enzyme to copy themselves. Certain HIV drugs work by these enzymes called "reverse transcriptases." Remarkably, Sedivy said, the reverse transcriptase of the major human retrotransposons called "L1" has been shown by researchers to be inhibited by some HIV drugs widely used in the clinic.
"The prospects of coming up with an existing drug therapy is something we really need to think about seriously," he said. "We're definitely going to test that and in the future, if needed, we also should be able to design new drugs that are highly specific for L1."
Ultimate success would provide a way to restore order in the cells and forestall at least some of the molecular ravages of age.
Provided by Brown University