Toward a new model of the cell: Everything you always wanted to know about genes

Turning vast amounts of genomic data into meaningful information about the cell is the great challenge of bioinformatics, with major implications for human biology and medicine. Researchers at the University of California, San Diego School of Medicine and colleagues have proposed a new method that creates a computational model of the cell from large networks of gene and protein interactions, discovering how genes and proteins connect to form higher-level cellular machinery.
The findings are published in the December 16 advance online publication of Nature Biotechnology.
"Our method creates ontology, or a specification of all the major players in the cell and the relationships between them," said first author Janusz Dutkowski, PhD, postdoctoral researcher in the UC San Diego Department of Medicine. It uses knowledge about how genes and proteins interact with each other and automatically organizes this information to form a comprehensive catalog of gene functions, cellular components, and processes.
"What's new about our ontology is that it is created automatically from large datasets. In this way, we see not only what is already known, but also potentially new biological components and processes – the bases for new hypotheses," said Dutkowski.
Originally devised by philosophers attempting to explain the nature of existence, ontologies are now broadly used to encapsulate everything known about a subject in a hierarchy of terms and relationships. Intelligent information systems, such as iPhone's Siri, are built on ontologies to enable reasoning about the real world. Ontologies are also used by scientists to structure knowledge about subjects like taxonomy, anatomy and development, bioactive compounds, disease and clinical diagnosis.
A Gene Ontology (GO) exists as well, constructed over the last decade through a joint effort of hundreds of scientists. It is considered the gold standard for understanding cell structure and gene function, containing 34,765 terms and 64,635 hierarchical relations annotating genes from more than 80 species.
"GO is very influential in biology and bioinformatics, but it is also incomplete and hard to update based on new data," said senior author Trey Ideker, PhD, chief of the Division of Genetics in the School of Medicine and professor of bioengineering in UC San Diego's Jacobs School of Engineering.
"This is expert knowledge based upon the work of many people over many, many years," said Ideker, who is also principal investigator of the National Resource for Network Biology, based at UC San Diego. "A fundamental problem is consistency. People do things in different ways, and that impacts what findings are incorporated into GO and how they relate to other findings. The approach we have proposed is a more objective way to determine what's known and uncover what's new."
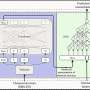
In their paper, Dutkowski, Ideker and colleagues capitalized upon the growing power and utility of new technologies like high-throughput assays and bioinformatics to create elaborately detailed datasets describing complex biological networks. To test the approach, the scientists pulled together multiple such datasets, applied their method, and then compared the resulting "network-extracted ontology" to the existing GO.
They found that their ontology captured the majority of known cellular components, plus many additional terms and relationships, which subsequently triggered updates of the existing GO.
Neither Ideker nor Dutkowski say the new approach is intended to replace the current GO. Rather, they envision it as complementary high-tech model that identifies both known and uncharacterized biological components derived directly from data, something the current GO does not do well. Moreover, they note a network-extracted ontology can be continuously updated and refined with every new dataset, moving scientists closer to the complete model of the cell.
Journal information: Nature Biotechnology
Provided by University of California - San Diego