From beaker to bits: Collaboration creates computational model of human tissue
Computer scientists and biologists in the Data Science Research Center at Rensselaer Polytechnic Institute have developed a rare collaboration between the two very different fields to pick apart a fundamental roadblock to progress in modern medicine. Their unique partnership has uncovered a new computational model called "cell graphs" that links the structure of human tissue to its corresponding biological function. The tool is a promising step in the effort to bring the power of computational science together with traditional biology to the fight against human diseases such as cancer.
The discovery follows a more than six-year collaboration, breaking ground in both fields. The work will serve as a new method to understand and predict relationships between the cells and tissues in the human body, which is essential to detect, diagnose, and treat human disease. It also serves as an important reminder of the power of collaboration in the scientific process.
The new research led by Professor of Biology George Plopper and Professor of Computer Science Bulent Yener is published in the March 30, 2012, edition of the journal PLoS One in a paper titled, "Coupled Analysis of in Vitro and Histology Tissue Samples to Quantify Structure-Function Relationship." They were joined in the research by Evrim Acar a graduate student at Rensselaer in Yener's lab currently at the University of Copenhagen. The research is funded by the National Institutes of Health and the Villum Foundation.
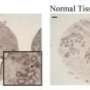
The new, purely computational tool models the relationship between the structure and function of different tissues in body. As an example of this process, the new paper analyzes the structure and function of healthy and cancerous brain, breast, and bone tissues. The model can be used to determine computationally whether a tissue sample is cancerous or not, rather than relying on the human eye as is currently done by pathologists around the world each day. The objective technique can be used to eliminate differences of opinion between doctors and as a training tool for new cancer pathologists, according to Yener and Plopper. The tool also helps fill an important gap in biological knowledge, they said.
Every budding biologist is taught a central concept: there is a relationship between biological structures like cells or organs and their functions. This structure/function relationship is well understood at some levels. For example, we know what individual brain cells are structurally comprised of and the functions they perform in the body. It is when you delve deeper into interactions between millions of different types of cells and dozens of specialized organs that our limited understanding of human biology is quickly brought to light.
"Previous biological analysis techniques simply ground up tissues and looked at things like gene expression. This is like looking at poll numbers after a vote. You don't know who voted for what or why, you only know who won the race," Plopper said. "With this new analysis we can explain how individual cells function in the system while still focusing on the entire system as a whole."
The ability to understand the complex relationship between cells in our tissues required an entirely new way to do biology, according to the scientists.
"We needed to take biology out of the Petri dish and microscope slide and lift it into a different space. We needed to integrate the interactions in the body on multiple scales," Yener said.
Yener and his computational counterparts did this by building a computational model using graph theory that linked several different biological data sets from Plopper and his biology counterparts. The biological data came from tissue samples physically removed from the human body and samples grown outside the body in the lab.
In the diagnosis of cancer, a pathologist removes a section of suspicious-looking tissue from a patient. The tissue sample is then sliced and stained for easier viewing and looked at through the age-old tool of biology, the microscope. This process is called histology. The pathologist is literally looking for cancer. When looking through the microscope, trained pathologists simply know cancer when they see it. They don't know how it came to be or where it is going, just that it is present in the tissue in front of them. In other words, they can see the structure of the cancer, but not know its function. In order to truly know the roots of diseases like cancer, scientists need to know how it arose in the tissue in the first place.
One way of seeing how cancer functions is to literally grow and watch cancerous tissue form in the lab. This process of growing cells in something like a test tube outside of the body is called cell culturing. It is a way to simplify and control the elaborate symphony of what is going on within the body, outside the body. But, a cell culture is only one small part of the symphony of our bodies. It can never tell the whole story.
Therefore, tools like histology and cell culture needed to be combined to get a fuller picture of the transition from healthy cells to full-fledged cancer. But no laboratory experiments exist to make the connection between the two. This is where computation can play a significant role, according to Plopper and Yener. Through computation Plopper and Yener were able to, for the first time ever, quantitatively compare histology samples with lab cultures.
The new computational method uses graph theory. Instead of looking at tissue as a series of cells, graph theory simplifies the system into a series of dots and lines with the dots being the cells and the lines their interactions. The program links and compares the accuracy of data from actual human tissue samples from histology with those grown in the controlled laboratory setting. The result is a new tool that can detect and distinguish cancer and quantify the actual differences between different tissues analyzed.
"Currently, biology is a lot of trial and error," Yener said. "With a mathematical model of something like cancerous tissue, you can computationally represent the relationships between cells. That model can then be predictive and eliminate time and energy lost in trial and error."
To non-scientists, interactions between two different types of scientists seems like the norm. But, there are no two groups of scientists more disparate than biologists and computer scientists, according to both Yener and Plopper. For biologists, discovery is in the details. How many genes are being expressed in a tissue? What protein is involved in the disease? Computer scientists seek to represent the larger relationship between different data points. What those data points actually represent is largely irrelevant.
"We have a very unique relationship," Plopper said. "I think it will take my whole career to get people in both fields who currently don't talk to even accept the fact that they should. But if all scientists understand how to generate and use numeric data of some sort, that would really lower the barriers between fields."
Plopper has even created a class, "Cell-Extracellular Matrix Interactions," to develop a new type of data-savvy biologist.
"These discoveries have to come from both fields," Yener said. "Scientists need to trust computational methodologies and that trust will only come when we go back to the basic biology that can be fed into the modeling and tested."
Provided by Rensselaer Polytechnic Institute