This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
proofread
A new deep-learning-based analysis toolkit for spatial transcriptomics
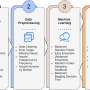
 graph and the differential evolution algorithm to transform slices and construct a stacked 3D alignment of a tissue. Credit: Nature Communications (2023). DOI: 10.1038/s41467-023-43220-3")
Biology and medical researchers use spatial transcriptomics (ST) technologies to detect transcription levels in cells, predict cell types and build a tissue's three-dimensional (3D) structure. However, this analysis can be difficult when there are multiple tissue slices that need to be analyzed jointly using state-of-the-art toolkits. It is challenging for researchers to assemble the slices and build the 3D structure manually.
To overcome this problem, a research team led by Prof. Qu Kun from the University of Science and Technology (USTC) of the Chinese Academy of Sciences (CAS) developed a new spatial architecture characterization by deep learning (SPACEL). Through three modules, Spoint, Splane, and Scube, SPACEL can build the 3D panorama of tissues automatically.
Their research results are published in Nature Communications.
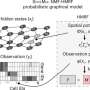
The three modules are designed for three main tasks in ST analysis. Sprint can perform cell-type deconvolution to predict the spatial distribution of the cell types. A combination of simulated pseudo-spots, neural network modeling, and statistical recovery of expression profiles ensure the robustness and accuracy of the prediction.
Splane employs a graph convolutional network approach and an adversarial learning algorithm to identify special domains by jointly analyzing multiple ST slices, while Scube automatically aligns the slices and constructs a stacked 3D architecture of the tissue. Through three modules, the 3D architecture of the tissue is built from the raw data.
Researchers applied SPACEL to 11 ST datasets, totaling 156 slices, and technologies like 10X Visium, STARmap, MERFISH, Stereo-seq, and Spatial Transcriptomics were involved during the process; SPACEL has demonstrated its superior performance over the others for cell type deconvolution in three core analytical tasks: predicting cell type distribution, identifying spatial domains, and reconstructing three-dimensional tissue structures.
This research provides a valuable integrated toolkit for ST data processing and analysis, benefiting further research employing ST technologies.
More information: Hao Xu et al, SPACEL: deep learning-based characterization of spatial transcriptome architectures, Nature Communications (2023). DOI: 10.1038/s41467-023-43220-3
Journal information: Nature Communications
Provided by University of Science and Technology of China