This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
proofread
Structure-destabilizing mutations transform Bcl-2 from an antiapoptotic protein into a proapoptotic protein

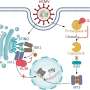
Bcl-2 family proteins are highly conserved molecules that play a crucial role in regulating the release of apoptotic proteins from mitochondria. They possess Bcl-2 homology (BH) domains, which are characterized by both sequence and structural similarities, and are essential for their interactions and functions.
The antiapoptotic members of this family, including Bcl-2, Bcl-XL, and Mcl-1, feature four BH domains (BH1 to BH4). On the other hand, proapoptotic multi-BH members like Bax, Bak, and Bok also have four BH domains.
In contrast, BH3-only members such as Bid and Bim only possess the BH3 domain. Some BH3-only proteins have the ability to directly activate Bax and Bak, leading to their homo-oligomerization and the formation of pores in the mitochondrial outer membrane (MOM). This process is critical in regulating apoptosis.
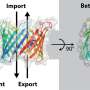
Surprisingly, the structures of multi-BH anti and proapoptotic proteins exhibit remarkable similarities. The main question lies in the possibility to transform anti-apoptotic Bcl-2 into pro-apoptotic proteins resembling Bax by modifying key structural elements within Bcl-2.
To address that, a group of researchers from China and the U.S. elucidates the mechanism of apoptosis induced by structural instability mutations in the mitochondrial protein Bcl-2.
The researchers conducted mutations on critical amino acids within a highly-conserved domain of the Bcl-2 protein. They found that glutamate at position 152 (E152) in the Bcl-2 protein plays a pivotal role in regulating its functionality. Mutations at E152 (E152A, E152C, E152S) were able to induce apoptosis by liberating cytochrome c from mitochondria in cells lacking Bax and Bak.
"Interestingly, when the transmembrane region of the Bcl-2 protein was excised, and E152 mutations were introduced, a multimeric structure formed in the mitochondrial outer membrane. This structural change exposed more BH3 domains, ultimately resulting in a pro-apoptotic function," said Jialing Lin, corresponding of the study published in Mitochondrial Communications.
Further mechanism studies showed that the mutation of E152 could eliminate the formation of hydrogen bonds with K22 and S105, thus reducing the structural stability of Bcl-2, and the mutants of K22 and S105 also had pro-apoptotic functions similar to that of the mutant E152. Consequently, through in vitro liposome experiments, the researchers found that both WT Bcl-2 and E152S mutant proteins can form large pores mediated by tBid, which regulated the release of molecules about 10kd in the membrane.
"Our study contributes to the existing literature on apoptosis," says co-corresponding author Quan Chen. "It introduces fresh concepts and potential avenues for the development of adjuvant drugs targeting Bcl-2 as a cancer treatment approach."
More information: Ping Gao et al, Structure-destabilizing mutations unleash an intrinsic perforation activity of antiapoptotic Bcl-2 in the mitochondrial membrane enabling apoptotic cell death, Mitochondrial Communications (2023). DOI: 10.1016/j.mitoco.2023.08.001
Provided by KeAi Communications Co.