PC deficiency suppresses seipin: New insights into Berardinelli-Seip congenital lipodystrophy type 2
. Credit: Life Metabolism (2022). DOI: 10.1093/lifemeta/loac021")
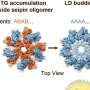
Berardinelli-Seip congenital lipodystrophy type 2 (BSCL2), the most severe form of lipodystrophy that leads to loss of nearly all subcutaneous fat tissue, is caused by mutations in the BSCL2/seipin gene. Seipin is an integral endoplasmic reticulum (ER) protein and has been shown to play important roles in lipid droplet homeostasis and lipid storage.
In seipin-deficient cells, defective lipid droplet biogenesis causes abnormal partition of lipids along with aberrant supersized lipid droplets, which is also reported in phosphatidylcholine (PC)-deficient cells. Besides the cellular lipid droplet defect, seipin deficiency also causes many physiological defects, such as fatty liver, diabetes, mental retardation and sperm abnormality. It is unclear whether all these defects are due to defective lipid droplet homeostasis or other mechanisms.
A study performed by scientists from the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, demonstrates that deficiency in the B12-one-carbon cycle-phosphatidylcholine (PC) axis ameliorates embryonic lethality but exacerbates lipid droplet defect in seipin mutants of the nematode C. elegans. This finding indicates that some of the physiological defects of BSCL2 may be independent of lipid droplet abnormality. The research is published in Life Metabolism.
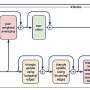
In this study, the authors used C. elegans BSCL2 model seip-1. seip-1 mutants exhibit both embryonic lethality and lipid droplet abnormality. To understand how seip-1 affects embryogenesis, they carried out a genetic screen to identify suppressors of the embryonic lethal phenotype in the seip-1 mutants. They identified transcription factor nhr-114 and spin-4, a putative transporter of the major facilitator family, as suppressors of seip-1 embryonic lethality. Both nhr-114 and spin-4 act in the same B12-one carbon cycle-PC pathway.
PC levels were significantly decreased by mutations of nhr-114 and spin-4 in the seip-1 mutants. These results indicated that mutations of nhr-114 and spin-4 suppress seip-1 embryonic lethality via PC deficiency. Consistently, mutation of pcyt-1, encoding a rate limiting enzyme in PC synthesis, also suppresses the embryonic lethality of the seip-1 mutants.
To explore the underlying mechanism of the suppression of PC deficiency on seip-1 embryonic lethality, the authors further examined the genetic interaction between seip-1 and a panel of flippases and scramblases, which shapes membrane phospholipid bilayer.
Intriguingly, knockdown of a C. elegans homolog of human VMP1, an ER-resident phospholipid scramblase, ameliorated the embryonic lethality of seip-1 and enhanced the suppressive effect of the suppressors. These results suggest that ER phospholipid homeostasis is important for the embryonic development of the seip-1 mutants.
To delineate the relationship between the roles of seipin in cellular lipid droplet homeostasis and physiological function, the authors examined the effect of PC deficiency on lipid droplet homeostasis in the seip-1 mutants. Remarkably, PC-deficient mutations further increased the number of supersized lipid droplets in the seip-1 mutants.
Put together, while PC deficiency suppresses the embryonic lethality, it exacerbates the large lipid droplet phenotype in the seip-1 mutants. These results indicate that seipin may regulate embryogenesis and lipid droplet homeostasis through distinct mechanisms.
The suppression of the physiological phenotype of seipin deficiency by PC reduction is somewhat unexpected, since both lead to a similar cellular lipid droplet phenotype. Examining the rescuing effect of reducing PC synthesis or VMP1 activity in vertebrate BSCL2 models will be the next target. This study also highlights the importance of distinguishing cellular phenotypes and physiological phenotypes in disease models.
More information: Jinglin Zhu et al, Reduced phosphatidylcholine synthesis suppresses the embryonic lethality of seipin deficiency, Life Metabolism (2022). DOI: 10.1093/lifemeta/loac021
Provided by Higher Education Press