Study reveals genetic diversity of a particularly problematic pathogen

Researchers at University of California San Diego School of Medicine and Jacobs School of Engineering, with colleagues at Baylor College of Medicine, have used a systems biology approach to parse the genetic diversity of Clostridioides difficile, a particularly problematic pathogen in health care settings.
The Centers for Disease Control estimates that the bacterium causes approximately 500,000 infections in the United States annually, with severe diarrhea and colitis (inflammation of the colon) as characteristic symptoms.
The researchers' findings are published in the April 27, 2022 online issue of PNAS.
C. difficile is the most dominant cause of hospital-associated infections, in part from the use of antibiotics, which can kill enough healthy bacteria to allow C. difficile to grow unchecked. Infections are particularly dangerous in older persons. One in 11 people over the age of 65 who are diagnosed with a hospital-associated case of C. difficile die within one month, reports the CDC.
"C. diff is persistent and pervasive," said senior author Jonathan M. Monk, Ph.D., a research scientist in the Systems Biology Research Group at UC San Diego, directed by Bernhard O. Palsson, Ph.D., professor of bioengineering and an adjunct professor in the UC San Diego School of Medicine. "It doesn't cause typical diarrhea. Most people do recover, but some become seriously ill, require hospitalization and some die from complications like kidney failure or sepsis."
To better understand the genetic features of C. difficile—and thus develop models that can identify and predict its complex and constant evolution—researchers used whole-genome sequencing, high-throughput phenotypic screening and metabolic modeling of 451 bacterial strains.
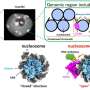
This data was used to construct a "pangenome" or entire set of genes representative of all known C. difficile strains, from which they identified 9,924 distinct gene clusters, of which 2,899 were considered to be core (found in all strains) while 7,025 were "accessory" (present in some strains but missing in others).
Using a new typing method, they categorized 176 genetically distinct groups of strains.
"Typing by accessory genome allows for the discovery of newly acquired genes in genomes of pathogens that may otherwise go unnoticed with standard typing methods," said co-author Jennifer K. Spinler, Ph.D., an instructor in pathology and immunology at the Baylor College of Medicine. "This could be critical in understanding what drives an outbreak and how to fight its spread."
Thirty-five strains representing the overall set were experimentally profiled with 95 different nutrient sources, revealing 26 distinct growth profiles. The team then built 451 strain-specific genome scale models of metabolism to computationally produce phenotype diversity in 28,864 unique conditions. The models were able to correctly predict growth in 76 percent of measured cases.
"One of the strengths of the presented work is the cohesion of distinct biological data types into comprehensive systems biology frameworks that enable analysis at scale," said first author Charles J. Norsigian, Ph.D., a data scientist in the Systems Biology Research Group. "By interpreting strains of C. difficile in a population context, we were able to bring to light pertinent strain features regarding nutrient niche, virulence factors, and antimicrobial resistance determinants that might have otherwise gone undetected."
Co-authors include Bernhard O. Palsson, UC San Diego; Heather A. Danhof, Colleen K. Brand, Firas S. Midani, Robert A. Britton and Tor C. Savidge, Baylor College of Medicine; and Jared T. Broddrick and Jennifer K. Spinier, NASA Ames Research Center.
More information: Charles J. Norsigian et al, Systems biology approach to functionally assess the Clostridioides difficile pangenome reveals genetic diversity with discriminatory power, Proceedings of the National Academy of Sciences (2022). DOI: 10.1073/pnas.2119396119
Journal information: Proceedings of the National Academy of Sciences
Provided by University of California - San Diego