New technology enables unprecedented glimpse inside single brain cells

Salk Institute researchers have developed a new genomic technology to simultaneously analyze the DNA, RNA and chromatin—a combination of DNA and protein—from a single cell. The method, which took five years to develop, is an important step forward for large collaborations where multiple teams are working simultaneously to classify thousands of new cell types. The new technology, published in Cell Genomics on March 9, 2022, will help streamline analyses.
"This multimodal platform is going to be useful by providing a comprehensive database that can be used by the groups trying to integrate their single-modality data," says Joseph Ecker, director of the Genomic Analysis Laboratory at Salk, the Salk International Council Chair in Genetics and Howard Hughes Medical Institute Investigator. "This new information can also inform and guide future cell-type classification."
Ecker believes this technology will be vital for large-scale efforts, such as the National Institutes of Health's BRAIN Initiative Cell Census Network, which he co-chairs. A major effort of the BRAIN Initiative is to develop catalogues of mouse and human brain cell types. This information can then be used to better understand how the brain grows and develops, as well as the role different cell types play in neurodegenerative diseases, such as Alzheimer's.
Current single-cell technology works by extracting either DNA, RNA or chromatin from a cell's nucleus, and then analyzing its molecular structure for patterns. However, this method destroys the cell in the process, requiring researchers to rely on computational algorithms to analyze more than one of these components per cell or to compare the results.
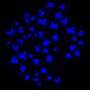
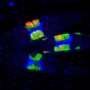
For the new method, called snmCAT-seq, scientists used biomarkers to tag DNA, RNA and chromatin without removing them from the cell. This allowed the researchers to measure all three types of molecular information in the same cell. The scientists then used this method to identify 63 cell types in the frontal cortex region of the human brain and benchmarked the efficacy of computational methods for integrating multiple single-cell technologies. The team found the computational methods have high accuracy in characterizing broadly defined brain-cell populations but show significant ambiguity in analyzing finely defined cell types, suggesting the necessity to define cell types by diverse measurements for more accurate classification.
The technology could also be used to better understand how genes and cells interact to cause neurodegenerative diseases.
"These diseases can broadly affect many cell types. But there could be certain cell populations that are particularly vulnerable," says co-first author Chongyuan Luo, assistant professor of human genetics at the David Geffen School of Medicine at UCLA. "Genetic research has pinpointed the regions of the genome that are relevant for diseases like Alzheimer's. We're providing another data dimension and identifying the cell types affected by these genomic regions."
As a next step, the team plans to use the new platform to survey other areas of the brain, and to compare cells from healthy human brains with those from brains affected by Alzheimer's and other neurodegenerative diseases.
More information: Joseph R. Ecker, Single nucleus multi-omics identifies human cortical cell regulatory genome diversity, Cell Genomics (2022). DOI: 10.1016/j.xgen.2022.100107. www.cell.com/cell-genomics/ful … 2666-979X(22)00027-1
Provided by Salk Institute