AI methods of analysing social networks find new cell types in tissue

In situ sequencing enables gene activity inside body tissues to be depicted in microscope images. To facilitate interpretation of the vast quantities of information generated, Uppsala University researchers have now developed an entirely new method of image analysis. Based on algorithms used in artificial intelligence, the method was originally devised to enhance understanding of social networks. The researchers' study is published in The FEBS Journal.
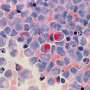
The tissue composing our organs consists of trillions of cells with various functions. All the cells in an individual contain the same genes (DNA) in their nuclei. Gene expression occurs by means of "messenger RNA" (mRNA)—molecules that carry messages from the nucleus to the rest of the cell, to direct its activities. The mRNA combination thus defines the function and identity of every cell.
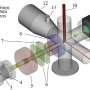
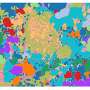
RNA transcripts are obtainable through in situ sequencing. The researchers behind the new study had previously been involved in developing this method, which shows millions of detected mRNA sequences as dots in microscope images of the tissue. The problem is that distinguishing all the important details may be difficult. This is where the new AI-based method may come in useful, since it allows unsupervised detection of cell types as well as detection of functions within an individual cell and of interactions among cells.
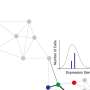
"We're using the latest AI methods—specifically, graph neural networks, developed to analyze social networks; and adapting them to understand biological patterns and successive variation in tissue samples. The cells are comparable to social groupings that can be defined according to the activities they share in their social networks like Twitter, sharing their Google search results or TV recommendations," says Carolina Wählby, professor of quantitative microscopy at the Department of Information Technology, Uppsala University.
Earlier analytical methods of this type of data depend on knowing which cell types the tissue contains, and identifying the cell nuclei in it, in advance. The method conventionally used, known as "single-cell analysis," may lose some mRNA and miss certain cell types. Even with advanced automated image analysis, it is often difficult to find the various cell nuclei if, for example, the cells are packed densely together.
"With our analysis, which we call 'spage2vec,' we can now get corresponding results without any previous knowledge of expected cell types. And what's more, we can find new cell types and intra- or intercellular functions in tissue," Wählby says.
The research group are now working further on its analytical method by investigating differentiation and organization of various types of cells during the early development of the heart. This is pure basic research, intended to provide more knowledge of the mechanisms that govern development, both when everything is functioning as it should and when a disease is present. In another project, a collaboration with cancer researchers, the Uppsala group are hoping to be able to apply the new methods to gain a better understanding of how tumor tissue interacts, at molecular level, with surrounding healthy tissue. The aim is that, in the long term, this will culminate in better treatments that can be adapted to individual patients.
More information: Gabriele Partel et al. Spage2vec: Unsupervised representation of localized spatial gene expression signatures, The FEBS Journal (2020). DOI: 10.1111/febs.15572
Provided by Uppsala University