Epigenetics: Inheritance of epigenetic markers

A study undertaken by an international team led by Ludwig-Maximilians-Universitaet (LMU) in Munich molecular biologist Axel Imhof sheds new light on the mechanisms that control the establishment of epigenetic modifications on newly synthesized histones following cell division.
The classical genetic code is not the only code involved in the regulation of cell differentiation and behavior in multicellular organisms. The instructions encoded in the nucleotide sequence of the genomic DNA determine which sets of genes are expressed within a given cell type. Their selective expression thus defines the differences between a muscle cell and a nerve cell, for example. However, there is a second level of control that contributes to the regulation of patterns of gene expression. This is based on chemical modifications of DNA and of the histone proteins in which it is packed. This epigenetic code is now recognized as a vital part of the process responsible for the differentiation—and maintenance—of different cell types in higher organisms, although virtually all cells in an individual carry the same complement of genetic information.
However, unlike the replication of the DNA sequence itself, the transmission of epigenetic information during cell division is not well understood. Now, a team led by Axel Imhof at LMU's Biomedical Center, in collaboration with research groups based at the Helmholtz Zentrum München and in Denmark, has used a combination of theoretical modeling and experimentation to elucidate the mechanisms that mediate the establishment of epigenetic marks following cell division. The findings, which appear in the journal Cell Reports, provide deeper insights into the inheritance of epigenetic histone modifications.
In higher organisms, most of the DNA in cells is found in a condensed form known as chromatin, in which the DNA is wrapped around particles made of proteins known as histones. In chromatin, the functional state of any given gene is largely dependent on exactly how it is packaged. More specifically, chemical modification of histones modulates the accessibility of the DNA in chromatin, and thus controls whether the proteins required for gene expression can actually bind to the DNA. In order to ensure the stable transmission to daughter cells of the gene expression patterns that define the identities of the different cell types, it is crucial that chromatin states are maintained during cell division.
In the new study, Imhof and his colleagues focused on two specific modifications of histone H3—methylation of the lysines at positions 27 and 36 (K27me and K36me). The attachment of a methyl group (CH 3 ) to the histone alters its binding affinity for regulatory proteins and changes the degree of chromatin compaction. K27me is usually found on H3 in regions where genes are inactive, while K36me serves as a marker for active genes.
The crucial question addressed in the study was: What happens to these modifications during the course of cell division? Cell division is preceded by DNA replication, which doubles the amount of DNA that has to be packed—and thus requires the synthesis of new histones. However, freshly synthesized histones carry no epigenetic modifications. How then do cells ensure that the new histones acquire the correct pattern of modifications within the newly formed chromatin?
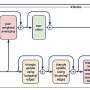
The problem is a tricky one, and the experimental approach adopted to solve it was technically challenging. The team first labelled newly synthesized histones with (non-radioactive) heavy isotopes. The new (heavy) histones could therefore be distinguished from the old (light) histones by means of high-resolution mass spectrometry. They then followed the fate of these two 'generations' of histones in the daughter cells after cell division.
The patterns of modification that they observed were extremely complex. In order to make sense of them, they devised two models for the inheritance of epigenetic histone modifications and used a computer-based procedure to compare the theoretical modification patterns with the dynamic changes detected in their labeling experiments. In theory, each of the lysines at positions 27 and 36 in histone H3 can be modified with one, two or three methyl groups. This meant that 16 possible isoforms had to be taken into consideration.
"Based on our modeling studies, we were able to demonstrate that the methylation patterns of the two functionally antagonistic residues K27me and K36me in cells reciprocally influence each other," says Axel Imhof. "The patterns that we actually observed can best be accounted for by the assumption that certain regions of the genome—which we refer to as domains—exhibit defined patterns of methylation." A further surprising finding was that, in rapidly dividing embryonal stem cells, the levels of demethylation observed during cell division were insignificant. The team now plans to investigate in greater detail what precisely is happening in these cells.
In the longer term, the researchers hope that their work will allow them to swiftly identify pathological alterations in the epigenetic states of cells. It is known that tumor cells often contain mutant forms of the enzymes responsible for the de novo modifications that occur during cell division, and this seems to be associated with the increased proliferation rates seen in such cells. "Consequently, a lot of work is currently going into the development of 'epidrugs' that could modulate the activity of these enzymes," says Imhof.
More information: Constance Alabert et al, Domain Model Explains Propagation Dynamics and Stability of Histone H3K27 and H3K36 Methylation Landscapes, Cell Reports (2020). DOI: 10.1016/j.celrep.2019.12.060
Journal information: Cell Reports
Provided by Ludwig Maximilian University of Munich