A complete set of energy level positions of all primary metal-halide perovskites

Metal-halide perovskites form a popular class of materials with intriguing optoelectronic properties. A fundamental understanding of the variations in the energy levels positions, as a function of the materials composition, is missing, however. Researchers from the TU/e and the University of Cologne have developed a new methodology to determine the absolute energy level positions of all primary perovskites, and provide explanations for the variations in these positions.
The materials class of halide perovskites (AMX3, where A is an alkali cation, or an organic cation, such as methylamine (MA) or formamidine (FA); B is lead or tin; X is a halide) has attracted enormous attention in the scientific community recently, due to breakthroughs in perovskite optoelectronics, mainly in photovoltaics and LEDs. By exchanging or mixing different ions in the perovskite crystal, it is possible to tune the optical gap of these semiconductors, allowing for an optimal overlap with the solar spectrum in absorption or a tunable wavelength of emission. The changes in band gaps are well characterized. However, the underlying physical origin of these changes, the shifts in the positions of the valence band maximum (VBM) and the conduction band minimum (CBM), are unknown. Knowing these positions is also crucial for designing contact layers that can inject/extract charge carriers efficiently into/from these perovskites, as is required in optoelectronic devices, or for designing multilayer heterojunction devices with proper band offsets between the layers.
"We were interested to understand the complex interplay of a few subtle yet correlated factors when combining different types of ions in the perovskite crystal structure," explains Shuxia Tao, Assis. Prof. from the Center of Computational Energy Research (CCER) of Applied Physics, TU/e. Together with Selina Olthof, experimental physicist from the University of Cologne, her team started about two years ago to address this problem, by initiating a large-scale experimental and theoretical investigation of all primary halide perovskites (18 materials in total).
-
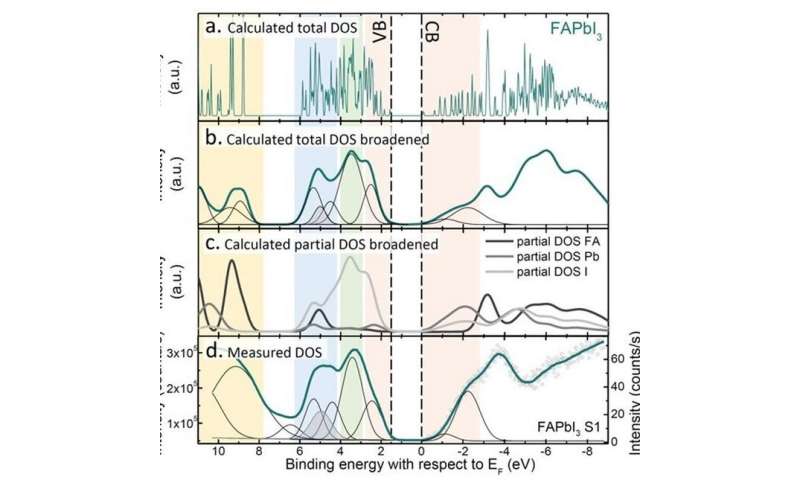
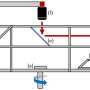
The developed methodology for determining the positions of the VBM and CBM by aligning measured UPS and IPES spectra with DFT calculated DOS. Credit: Eindhoven University of Technology -
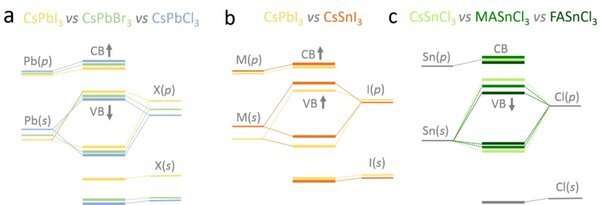
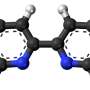
Schematic energy levels changes and their origin in AMX3 perovskites from tight binding analysis. Credit: Eindhoven University of Technology
The positions of VBM and CBM can be measured, in principle, by photoemission spectroscopy (PES), and inverse photoemission spectroscopy (IPES), respectively. Up till now, PES/IPES studies have reported quite diverse values for the VBM and CBM positions, however, even for common perovskite materials, because these positions are sensitive to variations in common data evaluation protocols.
Combining density functional theory (DFT) calculations and PES/IPES data, the researchers have developed a reliable methodology that is able to determine a consistent and accurate set of VBM and CBM data for all 18 perovskites. Further, by using a crystal orbital Hamiltonian population (COHP) analysis (Junke Jiang, Ph.D. candidate at CCER), and developing a tight-binding model (Prof. Geert Brocks, CCER), the researchers are able to explain the underlying origin of the trends observed in the perovskite energy level positions in terms of the energy levels of the individual cations and anions, and the hybridization between the corresponding atomic states.
"By combing several theoretical and experimental methods, we have created a new methodology that allows us to gain comprehensive insights of the electronic energy levels of this material class. We are very pleased with the outcome of this research after two years of continuous effort; we believe our work will make a broad impact in this field because this knowledge is crucial for further optimizing the perovskite materials as well as their energy alignment in a working device; both are very important aspects for the efficiency of the perovskite optoelectronic devices," adds Shuxia Tao.
Provided by Eindhoven University of Technology