Synthetic chemistry takes anti-cancer compounds out of the sea slug and into the lab

The natural world, with all its diversity, is a popular place for researchers to go looking for new drugs, including those that fight cancer.
But there is often a wide gap between finding a plant, sponge, or bacterium that contains a candidate drug, and actually bringing a medicine to the market. Maybe the compound gets flushed out of the human body too quickly to be effective. Or maybe it turns out you have to grind up a metric ton of farmed sea squirts just to get a single gram of the drug.
For that reason, it usually makes more sense to identify a compound with potential medicinal properties and then make it in the lab, instead of relying on organisms. Often, researchers look to the natural processes that create the compounds for inspiration as they develop synthetic analogs. Though this "biomimetic" method works, it has some limitations. For more than 10 years, Caltech's Brian Stoltz has been looking for a better approach, and now he has found it.
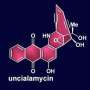
In December, Stoltz and his research team announced that they had developed a novel synthetic method for creating two compounds that hold the potential to become potent anti-cancer drugs. The compounds, jorumycin and jorunnamycin A, are naturally found only in the bodies of a black-and-white sea slug that lives in the Indian Ocean.
Both of those compounds are based around a backbone molecule known as bis-THIQ (bis-tetrahydroisoquinoline). In 40 years of research on bis-THIQ compounds, only one has been successfully brought into a clinical setting, Stoltz says. He hopes the production method developed in his lab can change that.
"We now have a synthesis that's going to let us make whole new compounds," he says. "It's going to enable us to do some really interesting drug-discovery research."
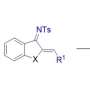
The production method is complex, involving the use of substances called transition metal catalysts, but essentially consists of adding hydrogen atoms to a simpler molecule in a series of steps. The addition of each hydrogen atom causes the molecule to fold further in on itself. When fully folded, the molecule is shaped in a way that makes it prone to bonding to and damaging DNA molecules. Medications that damage DNA might seem counterintuitive, but they are useful for targeting cancer cells. Since cancer cells multiply more quickly than healthy cells, they need to replicate their DNA more often, and are consequently much more sensitive to DNA damage.
Many compounds can damage DNA, but the trick is developing them into medications that are toxic enough to kill cancer cells, but not so harmful that they kill the healthy cells as well. The ideal medication will stay in the human body long enough to have a therapeutic effect, but not longer than about 24 hours.
Tailoring a compound to have the traits that make it an effective drug can be done by choosing what Stoltz calls "handles"—the various atoms and groups of atoms that stick off the molecular backbone. By choosing specific handles to put on a compound, researchers can give it the properties they desire.
This is where Stoltz's production method shines. Some handles interfere with biologically inspired syntheses of bis-THIQ compounds, but almost any handle will work with Stoltz's method, he says.
"It took us 10 years to get here, but now we can make new analogs in a week," he says.
Stoltz says Eric Welin, a postdoc on this research team, deserves much of the credit for refining the synthesis into an elegant solution.
"It was his creativity, drive, and decisiveness that pushed this forward," Stoltz says. "There was a way we could've finished this project that would've been a B-plus solution to the problem, but he pushed for the A-plus version. Eric insisted on developing a method that can produce either "left-handed" or "right-handed" versions of the final compounds at will, rather than the normal 50/50 mixture of both. It is a little like flipping a coin and being able to make it always land on heads."
He also credited another member of his research team, graduate student Aurapat "Fa" Ngamnithiporn, with doing much of the laboratory work necessary for performing the final synthesis, and continuing to produce novel non-natural analogs.
Further research will focus on using the synthesis to develop candidate drugs in collaboration with Dennis Slamon, an oncologist at UCLA.
The paper describing their findings, titled "Concise total syntheses of (–)-jorunnamycin A and (–)-jorumycin enabled by asymmetric catalysis," appears in the December 20 issue of Science.
More information: Eric R. Welin et al. Concise total syntheses of (–)-jorunnamycin A and (–)-jorumycin enabled by asymmetric catalysis, Science (2018). DOI: 10.1126/science.aav3421
Journal information: Science
Provided by California Institute of Technology