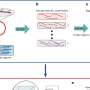
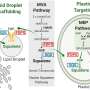
Complex networks identify genes for biofuel crops
 to discover genes that can be manipulated to enhance biofuels and bioproducts. The illustration shows how it works. High-resolution genomic data combined with co-expression datasets are used to build networks and then the networks are layered. Specific genes (orange) are identified, and a LOE score is calculated for each one (an example of the LOE score calculation is shown) that can be used to determine a set of potential target genes. Credit: US Department of Energy")
To improve biofuel production, scientists must understand the fundamental interactions that lead to the expression of key traits in plants and microbes. To understand these interactions, scientists are using different layers of information (about the relationships between genes, and between genes and phenotypes) combined with new computational approaches to integrate vast amounts of data in a modeling framework. Researchers can now identify genes controlling important traits to target biofuel and bioproduct production. The algorithm used in this work has been used to break the supercomputing exascale barrier for the first time anywhere in the world.
This approach lets scientists analyze massive data sets. They can do so using exascale computing, where computers perform 1018 calculations per second. With this approach, scientists can understand how cells work. They can use the insights to bioengineer beneficial traits into plants and microbes. The ability to use exascale computing opens up possibilities to study highly complex and interrelated molecular processes in cells at a level of detail not previously possible. Such computing also heralds a new era for systems biology.
Biological organisms are complex systems composed of functional networks of interacting molecules and macromolecules. Complex traits (phenotypes) within organisms are the result of orchestrated, hierarchical, heterogeneous collections of expressed genes. However, the effects of these genes and gene variants are the result of historic selective pressure and current environmental and epigenetic signals, and, as such, their co-occurrence can be seen as genome-wide correlations in different ways. Biomass recalcitrance (that is, the resistance of plants to degradation or deconstruction, which ultimately enables access to a plant's sugars for bioenergy purposes) is a complex multigene trait of high importance to biofuels initiatives.
To better understand the molecular interactions involved in recalcitrance and identify target genes involved in lignin biosynthesis/degradation, this study makes use of data derived from the re-sequenced genomes from over 800 different Populus trichocarpa genotypes in combination with metabolomics data (the concentrations of the metabolites) and pyrolysis-molecular beam mass spectrometry data. In addition, the scientists used other forms of gene regulation including co-expression, co-methylation, and co-evolution networks.
In analyzing this data, a team developed a "lines of evidence" (LOEs) scoring system to integrate the information in the different layers and quantify the number of LOEs linking genes to target functions. They applied this new scoring system to quantify the LOEs linking genes to lignin-related genes and phenotypes across the network layers. Applying the scoring system allowed for the generation of new hypotheses for new candidate genes involved in lignin biosynthesis in P. trichocarpa, including various AGAMOUS-LIKE genes (a type of transcription factor that controls the expression of other genes). The resulting Genome Wide Association Study networks are proving to be a powerful approach to determine the pleiotropic (genes that affect multiple phenotypes) and epistatic (multiple genes that work together to affect a single phenotype) relationships underlying cellular functions and, as such, the molecular basis for complex phenotypes, such as recalcitrance.
The algorithm in the CoMet software, which creates the co-evolution network used in this study, has since been ported to the new Summit supercomputer, currently world's fastest and smartest supercomputer at the Oak Ridge Leadership Computing facility. The research team used the CoMet software to break the exascale barrier, achieving a peak throughput of 1.88 exaops—faster than any previously reported science application—while analyzing genomic data on the Summit supercomputer. The research team achieved the feat, the equivalent to carrying out nearly 2 billion billion calculations per second, by using a mixture of numerical precisions on a new NVIDIA graphic processing unit computer chip technology called tensor cores. In this case, researchers implemented a new approach that used the tensor cores to obtain a dramatic increase in performance.
More information: Deborah Weighill et al. Pleiotropic and Epistatic Network-Based Discovery: Integrated Networks for Target Gene Discovery, Frontiers in Energy Research (2018). DOI: 10.3389/fenrg.2018.00030
Provided by US Department of Energy