Size matters... and structure too: New tool predicts the interaction of proteins with long non-coding RNAs
Far from just reading the information contained in the human genome, and in order to fully understand how it works, researchers aim to know the ins and outs of all the elements in this tiny regulated gear. Many laboratories, consortia and projects are devoted to get a global view of the functional regions of the genome and to know in which cell types genes are active.
Intriguingly, only a small fraction of the human genome (around 2%) contains genes encoding for proteins, which are the building blocks of the cell. The remaining 98% is important for regulation, meaning that it is involved in controlling when and where genes are active. This large portion of the genome produces RNA molecules, called non-coding RNAs, which differ in size, structure and function. As the different types of non-coding RNAs can interact with proteins in different ways, big efforts have been put into investigating them. Until now, there were no computational tools available to handle very long RNA sequences and studying them through experimental methods is at present a huge challenge.
In a recent article published in Nature Methods, researchers at the Centre for Genomic Regulation in Barcelona (Spain), in collaboration with scientists at EMBL's site in Monterotondo (Italy) and the California Institute of Technology (US), introduced a new computational tool to predict protein interactions with long non-coding RNAs, which they validated using advanced experimental techniques.
"Long non-coding RNAs interact with various proteins to mediate important cellular functions. Trying to identify these interactions can be a good starting point in order to understand the role of these molecules in the normal functioning of the cell but also in disease," explains Gian Gaetano Tartaglia, ICREA research professor at the Centre for Genomic Regulation (CRG) and principal investigator of this article.
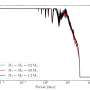
The new computational tool, which is called Global Score, allows scientists to predict where, along the sequence of a non-coding RNA, a protein will establish a physical contact. To do so, this algorithm integrates not only the global propensity of the protein to bind a particular RNA but also the local features of such a binding. "The structure of the RNA is absolutely important when predicting protein interactions. Our main challenge was to be able to work with RNA sequences regardless of their length in order to keep a complete view of their structural properties when looking for protein partners," adds Davide Cirillo, post-doctoral researcher at the CRG and first author of the paper. "The algorithm we have developed integrates this information and allows us not only to predict protein partners but also to prioritize them for experimental validation. This methodological advance will be crucial to better study long non-coding RNAs and their functions", concludes the researcher.
This work highlights, again, the relevant contribution of bioinformatics and computational biology to advance knowledge and their key role boosting and accelerating research in the life sciences.
More information: Davide Cirillo et al, Quantitative predictions of protein interactions with long noncoding RNAs, Nature Methods (2016). DOI: 10.1038/nmeth.4100
Journal information: Nature Methods
Provided by Center for Genomic Regulation