Signed, sealed, undelivered: Polycystic kidney disease develops when the message to generate cilia is 'lost in the mail'
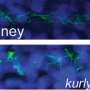
 compared with controls (left panel). Credit: Joshua Lipschutz, M.D., of the Medical University of South Carolina")
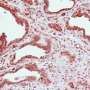
In an article published online ahead of print on Feb. 19, 2015 in the Journal of Biological Chemistry (JBC), investigators at the Medical University of South Carolina (MUSC) and the Ralph H. Johnson VA Medical Center report findings from in vitro and in vivo studies that elucidate the mechanisms underlying the impaired ciliogenesis and abnormal kidney development characteristic of polycystic kidney disease (PKD). Depletion of dynamin-binding protein or Tuba, a guanine nucleotide exchange factor, disrupted renal ciliogenesis in cell culture and led to abnormal kidney morphology in a Tuba knockdown zebrafish model of PKD.
Currently, no drug has been approved by the U.S. Food and Drug Administration to treat autosomal dominant PKD, which affects a half million Americans and more than 12 million people worldwide. The disease is characterized by the development of fluid-filled cysts in both kidneys, leading to end-stage renal disease, usually around age 50 to 60. In PKD, it is speculated that dysfunctional cilia are unable to detect the presence of urine flow, triggering reactivation of developmental pathways, which lead to the uncontrolled production of cysts that eventually destroy the kidney.
Cilia, the finger-like protrusions on most epithelial cells, were not so long ago thought to be as irrelevant to cell biology as the appendix is to physiology, a vestigial remnant of a long ago evolutionary past. Today, they are recognized as essential chemo-mechanical sensors that monitor and regulate what crosses into and out of a cell.
Dysfunctional cilia are now known to be implicated in not only PKD but a wide range of diseases affecting the eyes, ears, heart, and other organs. Understanding how cilia become dysfunctional in these diseases could provide insight into how to better treat or prevent them.
"How are cilia made? If you know that, you can figure out what goes wrong in ciliopathies, including polycystic kidney disease," says nephrologist Joshua H. Lipschutz, M.D., the senior author on the article, who holds a dual appointment at MUSC and the Ralph H. Johnson VA Medical Center.
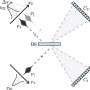
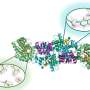
Much must go right for ciliogenesis to occur. Proteins necessary for ciliogenesis are manufactured in the endoplasmic reticulum before traveling to the trans-Golgi network to be sorted into "zip-coded packages" or vesicles for transport to the cilia. Lipschutz and others previously showed that the exocyst, a protein targeting complex, plays a crucial role in receiving these "zip-coded packages" containing ciliary proteins. The GTPase Cdc42 regulates the exocyst, which is the mailbox where these "packages" are received in the kidney. Renal ciliogenesis occurs only when the packaged proteins are delivered to the Cdc42-activated exocyst complex. Depleting either the exocyst or Cdc42 disrupts renal ciliogenesis.
In the JBC article, Lipschutz and his MUSC coauthors go a step further—showing in cell culture and a zebrafish model that depletion of Tuba, a guanine nucleotide exchange factor required for Cdc42 activation, also disrupts renal ciliogenesis. Tuba is thought to ensure that the Cdc42/exocyst mailbox is in place at the base of the cilia and ready to receive the packaged proteins. Without Tuba, Cdc42 is not appropriately activated, and the exocyst is mislocalized, so the undelivered packages continue to pile up, perhaps playing a role in the uncontrolled production of renal cysts in PKD.
When grown in a collagen gel, Madin-Darby canine kidney (MDCK) cells form into cysts, and the orientation of proteins, called polarity, are abnormal following Tuba knockdown. Specifically, apical proteins that would normally face the urinary space are mislocalized throughout the cell.
In zebrafish, injection of low doses of both Tuba and Cdc42 antisense morpholinos, which had no effect when administered separately, led to severe phenotypes similar to those seen following knockdown of other ciliary proteins. This is called genetic synergy and provides further evidence that both Tuba and Cdc42 are part of the same pathway. Because knockdown of Tuba in zebrafish affects cilia in a number of organs, including the brain, a variety of aberrant phenotypes were seen in the Tuba knockdown zebrafish model.
Lipschutz, who directs the zebrafish core at MUSC along with co-author Seok-Hyung Kim, Ph.D., is well aware of the advantages of the zebrafish for research—its genome is well characterized, it can be bred rapidly and inexpensively, and its transparent body enables easy visualization of aberrations under microscopy. However, the next step in this line of research will be to study the effects of Tuba depletion in the kidneys of mice, since murine kidneys are more like human kidneys than those of zebrafish.
Once the pathways underlying impaired ciliogenesis in PKD are more fully understood, therapeutic interventions can be designed to disrupt those pathways. As Lipschutz notes, "We do this research to help our patients. Further elucidating the pathways that underlie impaired ciliogenesis is an essential step in beginning to develop treatment options for PKD and other ciliopathies."
More information: Jeong-In Baek et al. Dynamin Binding Protein (Tuba) deficiency inhibits ciliogenesis and nephrogenesis in vitro and in vivo, Journal of Biological Chemistry (2016). DOI: 10.1074/jbc.M115.688663
Journal information: Journal of Biological Chemistry
Provided by Medical University of South Carolina