Scientists identify protein at death's door of cells

A protein embedded in the surface of mitochondria - the energy-producing batteries of living cells - opens the door to cell death, causing cells to experience severe power failures, according to new work by researchers at Temple University School of Medicine. The new study, appearing online September 17 in the journal Molecular Cell, suggests that blocking the door with a small molecule inhibitor could be key to the treatment of cardiovascular diseases such as heart attack and stroke, where extensive mitochondrial dysfunction and cell death hinder tissue recovery.
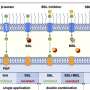
The study, led by Muniswamy Madesh, PhD, Associate Professor in the Department of Biochemistry, the Cardiovascular Research Center, and the Center for Translational Medicine at Temple University School of Medicine (TUSM), shows that the protein, spastic paraplegia 7 (SPG7), is the central component of the so-called permeability transition pore (PTP), a protein complex in the mitochondrial membrane that mediates necrotic cell death (death caused by cell injury).
The identification of SPG7 marks a major advance in scientists' understanding of how the PTP affects necrosis. Although first described in 1976, the molecular parts of the pore have eluded discovery. "The only known molecular component of the PTP prior to our discovery of SPG7 was a protein called CypD, which is necessary for pore function," Dr. Madesh explained.
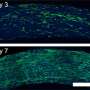
To identify genes that modulate PTP opening induced by calcium overload or increased levels of reactive oxygen species (ROS) - the two primary factors that cause mitochondrial dysfunction and cell death via pore opening - Dr. Madesh's team devised an RNA interference-based screen in which the activity of each gene under investigation was knocked down, or silenced, to examine its effects on mitochondrial calcium levels. The researchers began with a panel of 128 different genes but after initial screening narrowed the field to just 14 candidate PTP components. Subsequent experiments showed that the loss of only one of them - SPG7 - prevented pore opening.
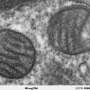
Much of what is known about the PTP comes from studies of mitochondria in disease. In pathological states, particularly those involving hypoxia (oxygen deficiency), calcium and ROS accumulate within mitochondria, causing them to swell and prompting the PTP to open. Because pore opening disrupts the flow of electrons and protons across the mitochondrial membranes, which normally sustains energy production, it results in a catastrophic drop in cellular energy levels.
In the absence of disease, precisely how the PTP helps to mediate normal cellular physiology remains unclear. According to Dr. Madesh, "Under physiological conditions, SPG7 may function through transient pore openings to release toxic metabolites that have accumulated in mitochondria." He plans to explore this possibility with knockout animal models.
Dr. Madesh also explained that the new findings could aid the development of novel therapeutics for conditions ranging from heart failure and stroke to cancer and neurodegeneration - all of which involve hypoxia and mitochondrial dysfunction to varying degrees. In these diseases, if the PTP could be prevented from opening, mitochondria could potentially continue to function, and cell death could be averted. With colleagues at TUSM, Dr. Madesh plans to explore the effects of SPG7 inhibitors in animals and, potentially, human patients.
Journal information: Molecular Cell
Provided by Temple University