New method helps map species' genetic heritage
Where did the songbird get its song? What branch of the bird family tree is closer to the flamingo - the heron or the sparrow?
These questions seem simple, but are actually difficult for geneticists to answer. A new, sophisticated statistical technique developed by researchers at the University of Illinois and the University of Texas at Austin can help researchers construct more accurate species trees detailing the lineage of genes and the relationships between species.
The method, called statistical binning, was used in the Avian Phylogenetics Project, the subject of a Dec. 12 special issue of the journal Science.
"A species tree is a way of describing how a species evolved from a common ancestor," said study leader Tandy Warnow, Founder Professor of bioengineering and computer science at the University of Illinois. "Researchers use a species tree to do all sorts of things, like figure out when different traits came into being, and what triggered that trait evolution, and how those things may or may not have been triggered by environmental changes."
There are two main approaches to constructing a species tree from genomic data, Warnow said. One method, which has prevailed for decades, puts all the gene data together into one giant matrix and analyzes it to map the overall species tree. This is called concatenation. The difficulty with that approach is that individual genes often have different lineages, which can diverge greatly from each other and the species tree as a whole.
A second approach, the coalescent-based method, looks at the data for each gene and estimates gene trees for each trait. Then it combines all the trees together to create the overall species tree. While this approach is sound theoretically and statistically, it does not perform as well as expected in practice.
"We realized that the gene trees that are combined have error in them," Warnow said. "When the gene trees have error, then when you combine them you get a bad estimate of the species tree. So we needed to get better gene trees, and the question is, how do we do that?"
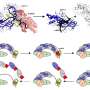
Statistical binning takes all the gene data and uses statistical optimization techniques to sort the genes into sets or "bins." The genes in each bin have trees that don't seem to have statistically significant differences. The data for each bin is combined into a "supergene" tree, and then the supergene trees are combined into an overall species tree.
"You can think of statistical binning as combining the best properties of the two dominant approaches," said Siavash Mirarab, graduate student at the University of Texas at Austin and first author of the paper detailing the statistical binning method. "Without this method, what people had to do was throw away data they didn't like. This approach allows you to use all the data you have and you don't have to throw away anything. We have a method that achieves that by grouping things together in a way that makes sense, statistically."
The researchers compared the species trees produced using the coalescent method with statistical binning to trees produced with concatenation or coalescence alone for several biological classes, such as birds, mammals, yeast and others. They found that adding the statistical binning process to the pipeline produced species trees that were better than the trees produced by either of the conventional methods.
"We sort the gene data in a sophisticated statistical way, but having done it we get better trees," Warnow said. "The result is significantly improved estimates of the gene trees, which gave us better estimates of the species tree and branch lengths, which helps you figure out when things happened. Everything was much more accurate."
Statistical binning allowed the Avian Phylogenetics Project to analyze more than 14,000 genes - one of the largest such projects yet published - and construct a large tree linking many different bird species. As it turns out, the flamingo is more closely related to the pigeon.
Warnow and Mirarab plan to continue to refine the statistical binning method and hope that it can add accuracy to many other similar studies.
"There's a large divide in the research community as to whether to use concatenation of a coalescent analyses. What we did was understand why the coalescent method didn't give good results and came up with a way of improving the input so that it could have good results. It's a way of bringing these two very divided communities into greater agreement with each other," Warnow said.
More information: Statistical binning enables an accurate coalescent-based estimation of the avian tree, www.sciencemag.org/lookup/doi/ … 1126/science.1250463
Journal information: Science
Provided by University of Illinois at Urbana-Champaign