Simulation method identifies materials for better batteries

(Phys.org) —Researchers from the University of Cambridge have devised a new simulation technique which reliably predicts the structure and behaviour of different materials, in order to accelerate the development of next-generation batteries for a wide range of applications.
The researchers, from the Cavendish Laboratory and the Department of Chemistry, are devising new methods for understanding the complex interactions that take place within lithium-ion batteries, in order to identify and develop the best materials to make better batteries.
Using software to predict the characteristics of materials before they're synthesised in order to guide and interpret experiments, the researchers successfully predicted the structures of a series of lithium silicides, an important step in understanding batteries made of silicon, and have also predicted new structures for a battery based on germanium. Details are published in the journal Physical Review B.
The lithium-ion battery market is large and growing rapidly. However, for many next-generation applications, such as longer-range electric vehicles and longer-lived consumer electronics, batteries which are smaller, more efficient and charge faster are needed.
Silicon has been proposed as a replacement for carbon in battery anodes, or positive electrodes, for the past 20 years. The anode must safely store lithium atoms when charged then release lithium ions, in turn causing electrons, which carry electrical current, to flow from the positive to the negative electrode. The more lithium the anode can store, the larger its capacity.
Most modern anodes are based on graphite, but silicon has roughly ten times the storage capacity, which in turn would greatly expand the capacity of the batteries used in mobile phones, electric vehicles and other applications. However, difficulty in managing silicon's properties has prevented the technology from being applied at scale.
"It's always exciting when we're able to successfully predict something, but it's especially gratifying to predict something that's really useful," said Dr Andrew Morris of the University's Cavendish Laboratory, the paper's lead author.
Lithium ion batteries are widely used due to their high energy density and high specific energy. They have a higher capacity than any other type of commercially-available battery, but in order to enable next-generation electronic devices, materials which can absorb, store and then release lithium quickly and safely need to be found.
More efficient batteries could potentially revolutionise a number of industries: for instance, enabling an electric car battery to be recharged in the same amount of time it currently takes to fill up a petrol tank, or allowing the energy generated by renewables to be stored for when it is most needed.
The various structures which the researchers predicted for lithium silicide have been shown to be stable in physical experiments, and further experiments are currently underway in Professor Clare Grey's laboratory in the Department of Chemistry to determine whether the same is true for the structures they predicted for germanium and to determine the roles these structures play on battery cycling.
The arrangement of atoms in a material determines its properties – what it looks like, how hard or soft it is, or what electrical state has. Performing simulations in a computer is far more efficient than making new materials in a laboratory and performing physical experiments on them. "What happens in a battery is complicated, and we need to work out what the model compounds should be," said Morris.
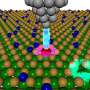
To perform their predictions, the researches used a method called AIRSS (Ab Initio Random Structure Searching), developed by one of the authors, Professor Chris Pickard of University College London, and Professor Richard Needs of the University's Cavendish Laboratory. Starting with a random collection of atoms, AIRSS is able to predict the crystal structure of the materials they will form using only quantum mechanics.
More information: Physical Review B, DOI: 10.1103/PhysRevB.90.054111
Journal information: Physical Review B
Provided by University of Cambridge