Molecular matchmaking for drug discovery
For millennia, mankind has discovered new drugs either through educated guesswork or blind luck. But with the proliferation of advanced computing, a new paradigm has emerged whereby one can find drug targets through simulation and modeling.
A combination of modeling, simulation, analysis and visualization, the process is accomplished through the expert application of biophysical algorithms and the high-performance, parallel-processing supercomputers of the Texas Advanced Computing Center's (TACC).
"Computers are a good way to accelerate the process of drug design," said Bajaj. "It takes 10 years to proof out a drug, and a billion dollars or more. Hence computational drug discovery is not only timesaving, but economics tells you this is the way we should be going."
[Recent studies suggest the cost and time required to move from the discovery phase to delivery is growing, with fewer drugs approved by the FDA per billion dollars spent. These findings highlighting the need for faster, cheaper methods.]
The work of discovering a breakthrough new drug begins with a careful analysis of the virus, bacteria or mutation that causes the illness. By shooting powerful x-rays through a sample, electron microscopes create nanoscale pictures of the relevant molecules in near-natural conditions. The images that emerge from these microscopes are a speckled mess, however, and must be cleaned up considerably to become useful.
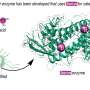
Combine 100,000 of these cleaned-up images and you have a three-dimensional model that can tell you about the structure of the molecule you are exploring. Structure is an important characteristic for drug discovery because it reflects the shape of the relevant molecules, and shape complementarity — the degree to which two molecules fit together — is a major factor in whether a drug will bond: the first step toward accomplishing its mission. When done correctly, the 3D model allows one to understand, identify and test potential binding sites on a virus.
"The nice thing about the electron projection is that you can build up volumetric models, so you can not only build a surface model from the outside, but also include information that is inside," Bajaj said.
As a consequence of improvements to the image reconstruction and modeling algorithms, Bajaj and his collaborators can now identify secondary structures of molecules, like individual side-chains — floppy but crucial limbs that extend from the central spine of molecule. This level of detail is required to accurately predict binding.
A recent paper by Bajaj, Samrat Goswami (Exa Corporation), and Qin Zhang (UT Austin) in the February 2012 edition of the Journal of Structural Biology showed that their algorithms were able to detect the secondary structures (α-helices and β-sheets) of proteins through intermediate (6-10 Å) and coarse (10-15 Å) resolution 3D maps reconstructed primarily from single particle cryo-electron microscopy.
"If you don't get all the factors into simulation, you get the wrong answer and your predictions suffer," said Bajaj. "And if your predictions suffer, you haven't done anything to accelerate the solution. You're just loading yourself down."
Once a target molecule's structure has been determined, it is then necessary to test potential drug compounds to see if any of them might fit a binding site. In the case of the HIV virus, as studied by Bajaj, the goal is find a molecule that can bind to a specific location on the virus' surface and signal to the virus that it has reached its destination. Rather than injecting its genetic material into a host cell, the potential drug would induce the virus to spill its contents in the extra-cellular medium where it would do no harm.
But in a world of potential compounds, how does a scientist find the one drug molecule that might match and bind to the target region?
Over the last decade, computer scientists have found faster ways to search for things using computers. Call it the "Googlization" of research. Bajaj has taken these insights and applied them to the problem of drug target screening. Instead of using search algorithms to find the closest coffee shop, Bajaj and his research team use them to create an ordered list of targets based on the binding energies and biochemical dynamics when two molecules come into contact, which indicates the most compatible compounds.
The molecules with the highest ranking are then visualized by Bajaj and analyzed to reveal where the algorithms have worked and where they can be improved.
"We not only quantify the accuracy, but also the errors that we make," Bajaj explained. "Without knowing the cause of error, how can you go back and improve your model?"
To make matters more difficult, researchers must ensure that the target compounds don't bind to other vital biological molecules, as these can lead to serious side effects. The sheer number of possible combinations and configurations boggles the mind. But as computer hardware and algorithms improve, such problems are becoming tractable.
Bajaj uses practically every system in TACC's computational ecosystem to solve these problems, including Ranger and Lonestar (TACC's high performance computing systems), Longhorn (TACC's remote visualization system) and Stallion (the super high-resolution tiled display in the TACC/ACES Visualization Laboratory).
"We are blessed at UT Austin with having all of these high performance computers and the TACC organization, because without them we couldn't make progress," Bajaj said.
Utilizing both CPU- and GPU-based methods, and mapping different processes to different architectures to accelerate the computation, Bajaj and his team improved the resolution and accuracy of the drug models tremendously. They have also sped up the docking search by an order of magnitude. "What used to take months is now taking a few days," he said.
According to J. Tinsley Oden, director of ICES, Bajaj's research has the potential to transform the way the drug discovery process works.
"Dr. Bajaj's breakthrough work on computer modeling the incredibly complex mechanisms central to the design, behavior, and delivery of drugs for combating infectious diseases is a testament to the power of modern computer modeling and simulation, high performance computing, computer visualization, and to Bajaj's special abilities and deep insight into both computer modeling and the biological processes involved in drug delivery," he said.
Computational drug discovery is a hot topic in academic research centers and industry alike. As chair of a study section of the National Institutes of Health, Bajaj often speaks with individuals from the pharmaceutical industry about changes in the field.
"More and more, they're moving into the computational drug screening arena, and more and more it's teams of people working together," Bajaj said. "The biophysicist, the biochemist and the synthetic chemist are sitting together with the computational expert, and they say it's giving them clues as to what they should be doing next."
Chandrajit Bajaj, professor of computer science at The University of Texas at Austin has been integrally involved in these efforts for more than 20 years. Computational and Applied Mathematics chair in Visualization and director of the Computational Visualization Center at UT's Institute for Computational Engineering and Sciences (ICES), over his career Bajaj has systematically attacked each step of the drug discovery process, improving the speed and accuracy of the algorithms involved in computational drug discovery. The research has been sponsored by the National Institute of Health, the National Science Foundation, and the Texas Institute of Drug and Diagnostic Development.
Provided by University of Texas at Austin, Texas Advanced Computing Center