New study reveals structure of the HIV protein shell
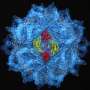
 form the core of the hexamer, and this core is surrounded by a floppy belt of C-terminal domains (colored blue). Credit: The Scripps Research Institute")
New research by scientists at The Scripps Research Institute and other institutions provides a close-up look at the cone-shaped shell that is the hallmark of human immunodeficiency virus (HIV), revealing how it is held together—and possible ways to break it apart.
Previously, scientists had known that the genetic material within HIV is enclosed within a shell called the capsid, which is formed by a honeycomb arrangement of about 250 hexagonal protein building blocks. For HIV to infect human cells, the virus binds to cell surface receptors, and then the capsid is delivered into the cytoplasm of the cell.
Now, in an advance, online issue of the journal Cell published on June 11, 2009, Professor Mark Yeager and colleagues at The Scripps Research Institute, the University of Virginia, and the University of Utah describethe first high-resolution molecular structure of the hexagonal protein building block, called CA, that makes up the HIV capsid. This detailed description may help scientists identify new ways to block HIV infection.
Bringing Down the Capsid
Since HIV/AIDS was first recognized in 1981, several drugs and drug combinations have allowed infected individuals to live longer and healthier lives. However, resistance to the existing drugs has created an urgent need for novel therapeutic strategies.
Current drugs target critical steps in the virus life cycle. For example, protease inhibitors block the protein cleavages that generate viral components—one of them being the protein CA.
Other possible ways to block infection would be to prevent formation of the capsid by blocking assembly of CA molecules or to find a way to disassemble the capsid once it is made.
"Anything that destabilizes the capsid, either by inhibiting assembly or accelerating disassembly should attenuate or even kill the virus," says Owen Pornillos, an investigator in Yeager's lab and first author of the Cell paper.
But to destabilize the capsid, it's necessary to know precisely how it is held together.
Making Crystals
"No one had been able to visualize the CA hexamer at atomic resolution," says Yeager. "Other groups had been able to solve structures of individual regions of CA. But it was not clear from these structures exactly how the CA proteins fit together."
To make the capsid, sets of six CA protein molecules first form hexamers, which then associate with one another to build a honeycomb-like shell comprised of about 250 hexamers. The ends of the shell are closed by insertion of seven and five CA protein pentamers, yielding the characteristic cone-like appearance of the capsid.
In 2007, Yeager's group managed to view the CA hexamers by a type of electron microscopy in which the samples are quick frozen in buffers, which preserves the inherent structure of proteins. That study provided the first glimpse of how CA proteins are arranged in the capsid. (The first author of the 2007 article was Barbie Ganser-Pornillos, Owen Pornillos' wife, who was also involved in the current study.)
In order to view the CA hexamer at even higher resolution, Yeager's group turned to X-ray crystallography. This technique requires growing 3D crystals of a molecule and then scattering a beam of X-rays off the crystals, which are recorded on a detector. Computational methods are then used to interpret the scattering patterns to calculate the position of every atom in the crystallized molecule.
But growing large, 3D crystals of the CA hexamer was no easy feat. The two ends of each CA protein molecule are held together by a "floppy" bridge, which precluded formation of orderly arrays of CA hexamers to form 3D crystals.
To overcome the problem, Pornillos and Yeager turned to molecular biology. They engineered CA proteins that would form sturdy chemical links between them, relying on the 2007 structure as their roadmap to determine exactly where to place the links.
"Our work takes advantage of so-called hybrid methods—molecular biology, biochemistry, electron microscopy, and X-ray crystallography," says Yeager. "These methods are synergistic. The EM results guided the molecular biology to engineer stable CA hexamers that were then amenable to 3D crystallization and X-ray structure analysis at atomic resolution."
The structure they obtained provided a view of the CA hexamer at an unprecedented resolution of two-Ĺngstrom (one Ĺngstrom equals one ten-billionth of a meter).
A Close-Up Look
All proteins are composed of linear chains of amino acids—with one end called the N-terminus and the opposite end the C-terminus—that are folded in three-dimensional shapes. In the CA protein, amino acid chains are twisted into several rods, called a-helices, with extensions—called side chains—that protrude from the main chain to interact with other folded regions of the protein.
The two-Ĺngstrom structure showed the positioning of these a-helices and, for the first time, the location of the atoms in the side chains. "We could precisely delineate all the chemical interactions that stabilize the hexamer," says Yeager.
The center of the CA hexamer is formed by 6 N-terminal ends of the CA protein subunits. The C-terminal domains form a "floppy" belt around this central core, connecting adjacent hexamers. The fact that the belt is not held rigidly in place, helps explain how the honeycomb shape of the capsid forms. "The curvature of the capsid is not constant," says Pornillos. "Now we can see in atomic detail how flexibility in CA makes this happen."
The group discovered another set of interactions critical to stabilizing the capsid—connections between the N-terminal and C-terminal ends of adjacent CA protein molecules in one hexamer. "Think of the fingers of one hand as the N-terminal domain and the palm as the C-terminal," says Yeager. "Imagine the fingers of one hand being cradled in the palm of the other, and so on as if you had six hands in a ring."
Knowing precisely how and where CA proteins interact gives researchers clues on how to interfere with these connections. One approach is to design small molecules that can insert themselves at strategic positions, impeding capsid assembly or making the capsid less stable.
While finding HIV therapies is a main driver for Yeager's work, he points out that it also provides fundamental insights into biology. "Determining the assembly of a relatively simple structure like the capsid of a virus can help us understand how more complex biological structures inside the cell are organized," he explains.
Source: The Scripps Research Institute (news : web)