Intrinsic changes in protein shape influence drug binding
Computational biologists at the University of Pittsburgh School of Medicine have shown that proteins have an intrinsic ability to change shape, and this is required for their biological activity. This shape-changing also allows the small molecules that are attracted to a given protein to select the structure that permits the best binding. That premise could help in drug discovery and in designing compounds that will have the most impact on protein function to better treat a host of diseases.
The findings were published this week in the online version of the Proceedings of the National Academy of Sciences.
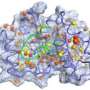
According to the classical view, known as "induced fit," drug binding causes a change in the target protein structure, explained senior author Ivet Bahar, Ph.D., professor and John K. Vries Chair of the Department of Computational Biology, Pitt School of Medicine. But it now appears that a protein has many different conformations that are already available even without the presence of a binding molecule, which is called the ligand. The ligand attaches to the protein shape that allows it to fit well, and that close interaction can lead to effective inhibition of protein function.
Gathering information about the array of conformations a target protein might exhibit can be of great use when designing new drugs, Dr. Bahar said. That allows the scientist to better identify the structural pocket into which the drug must fit to cause significant alterations in protein function, such as the inhibition of an enzyme reaction.
For the study, Dr. Bahar and her doctoral student, Ahmet Bakan, focused on three common drug targets, namely enzymes important in HIV, inflammatory response and the cell division cycle. Using the sets of conformations of protein-ligand complexes stored in the Protein Data Bank, an information repository for the scientific community at Rutgers University, the researchers figured out what structures the enzymes had both alone and when bound to a variety of small molecules.
"It seems there are simple but robust rules that control ligand binding," Dr. Bahar explained. "If we know the rules, we can make better predictions about which binding sites to target to make more effective drugs."
Source: University of Pittsburgh