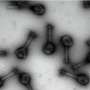
Enzyme helps cancer cells avoid genetic instability
 because they cant use homologous recombination to repair their DNA. But the cells can resume proliferation by cranking up their output of cathepsin L, which destroys 53BP1, a protein that prods the cells to use alternative, sloppier repair mechanisms. Credit: Susana Gonzalo")
Cancer cells are resourceful survivors with plenty of tricks for staying alive. Researchers have uncovered one of these stratagems, showing how cells lacking the tumor suppressor BRCA1 can resume one form of DNA repair, sparing themselves from stagnation or death. The study appears in the January 21st issue of The Journal of Cell Biology.
The BRCA1 protein helps to mend double-strand DNA breaks by promoting homologous recombination. Without it, cells can amass broken, jumbled, and fused chromosomes, which may cause them to stop growing or die. Although cells lacking BRCA1 seem like they should be vulnerable, loss of the protein instead seems to boost abnormal growth.
Recent studies have shown that cells lacking BRCA1 compensate by cutting back on 53BP1. This protein helps orchestrate a different DNA repair mechanism, nonhomologous end joining (NHEJ), and it thwarts a key step in homologous recombination. Researchers think that, in cells without BRCA1, 53BP1 spurs excessive NHEJ that can cause fatal chromosomal chaos. But with 53BP1 out of the way, the cells are able to resume homologous recombination. That might explain why cells that lack BRCA1 and eliminate 53BP1 can withstand traditional chemotherapy compounds and PARP inhibitors, a new generation of anti-cancer drugs that are in clinical trials. But how do cancer cells turn down 53BP1?
Researchers previously found that certain mutant fibroblasts increase production of cathepsin L, a protease that destroys 53BP1. BRCA1-deficient cancer cells take advantage of the same mechanism, according to a team of researchers led by Susana Gonzalo from the Washington University School of Medicine. When they cultured breast cancer cells that were missing BRCA1, the cells stopped growing. After two weeks of lethargy, however, some cells, which the researchers dubbed BOGA cells (BRCA1-deficient cells that overcome growth arrest), began to divide again. These cells showed increased levels of cathepsin L and reduced amounts of 53BP1. Eliminating cathepsin L from BOGA cells or dosing them with vitamin D, a cathepsin L inhibitor, prevented the decline in 53BP1 abundance.
To find out whether boosting cathepsin L levels enabled the cancer cells to restart homologous recombination, the researchers monitored sites of DNA damage tagged by RAD51, a protein that helps promote homologous recombination. The cells that had stopped growing did not display RAD51 foci, but these foci were prevalent in BOGA cells with reduced 53BP1. Removing cathepsin L from BOGA cells increased 53BP1 levels and diminished the number of RAD51 foci.
If cells can't perform homologous recombination, they turn to repair mechanisms such as NHEJ that can lead to jumbled chromosomes. However, after DNA-breaking doses of radiation, BOGA cells exhibited few chromosome defects. The number of these flaws climbed after the researchers stabilized 53BP1 levels by inhibiting cathepsin L or trimming its abundance.
The team then analyzed tumor samples from breast cancer patients. Researchers suspect that cathepsin L attacks 53BP1 by entering the nucleus. Samples from patients with BRCA1 mutations or with triple-negative breast cancer—an aggressive form of the disease—showed high levels of nuclear cathepsin L and reduced quantities of 53BP1. That suggests tumors in these patients hike the amounts of cathepsin L in the nucleus to break down 53BP1 and restore homologous recombination.
"It's a new pathway that explains how breast cancer cells lose 53BP1," says Gonzalo. How cancer cells boost nuclear cathepsin L levels is unclear, she notes.
Triple-negative breast cancers are currently identified by their lack of Her2 and the estrogen and progesterone receptors. The work suggests that another trio of measurements—the amounts of 53BP1, cathepsin L, and vitamin D receptor in the nucleus—might help identify patients that are resistant to current breast cancer treatments. These people might respond to cathepsin inhibitors, some of which are undergoing animal testing. These compounds might steer the cells away from homologous recombination and leave them vulnerable to other therapies.
More information: Grotsky, D.A., et al. 2013. J. Cell Biol. doi:10.1083/jcb.201204053
Journal information: Journal of Cell Biology
Provided by Rockefeller University