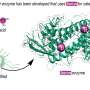
Structure, dynamics of a chemical signal that triggers metastatic cancer revealed
 are bound Credit: Riccardo Baron et al., UC San Diego")
In cancer and other pathological diseases, researchers are discovering that packaging is important: specifically, how DNA – about two meters long when unwound and stretched – coils up and compacts neatly inside the nucleus of a cell.
What they’ve learned is that molecular signals that control the packaging of DNA are critical to the activation and silencing of genes in the human body – a process generally described as epigenetics.
Now, a team of researchers from UC San Diego and the University of Pavia in Italy, with the help of high-performance computers housed at the San Diego Supercomputer Center (SDSC), have captured the chemical structure of one such signal – in static crystal form and in motion – which is at the heart of a variety of morphological events including the rapid movement of cells during embryonic development, wound healing, and cancer.
The results offer a potentially new path to combat metastatic cancer by blocking the activity of this epigenetic signal, which, among other things, has been shown to silence a gene responsible for cell-to-cell adhesion, a “molecular glue,” thus allowing cancer cells to spread.
“Our study opens the understanding of the molecular interaction and dynamics to be targeted to develop epigenetic drugs which hopefully will lead in the future to potent drugs against cancer,” said J. Andrew McCammon, Joseph Mayer Chair of Theoretical Chemistry and Professor of Pharmacology at UC San Diego and a Howard Hughes Medical Institute Investigator.
Historically, cancer researchers have generally focused on genetic mutations, specific changes in DNA which alter the function of the proteins they encode; studies ultimately have yielded several targeted drugs based on this approach. But treatments for many forms of cancer remain limited, prompting the search for other novel approaches. In particular, some have turned to epigenetics and processes that activate or silence genes by altering the physical structure of DNA -- how it’s packaged -- leaving its message or sequence intact.
“The full potential of epigenetic therapy is far from being exploited,” said Riccardo Baron, a postdoctoral researcher in McCammon’s lab and first author of the study, published in the February issue of the journal Structure. “Very little has been done in terms of pharmacological manipulation and studies such as this are a start down that road. Computer applications in chemistry hold great promises for designing new experiments and future drug development.”
Briefly, to compact an otherwise lengthy strand of DNA neatly inside the nucleus, cells rely on proteins called histones. DNA tightly loops around histones to form nucleosomes, the so-called “beads- on-a-string” that coil up to make up chromatin, the basic unit of chromosomes. Here, the DNA remains sequestered until it’s silenced or activated by enzymes responsible for gene expression.
One such enzyme coming under increasing scrutiny lately is lysine-specific demethylase 1, or LSD1 (no relation to the hallucinogen). Specifically, in 2004 researchers at Harvard University and University of Pavia found that LSD1 – particularly when bound to another protein called CoREST – removes one or more methyl groups from the amino acid lysine on histone H3, in a region that protrudes from the globular core known as the N-terminal tail. The result: DNA closes up shop, shutting down gene expression.
In a study published last year by a team at the University of Kentucky, it was discovered that the LSD1-CoRest complex worked in tandem with an enzyme called SNAIL1 to silence the activity of a gene responsible for E-cadherin, considered a type of “molecular glue” that keeps cells together. When this gene is repressed, cancer cells are allowed to spread – a hallmark of metastasis.
SNAIL1, a master regulator of the epithelial-mesenchymal transition (EMT) process that’s at the heart of many morphological events, has been found in high quantities in the sera of patients with several cancers, including breast and certain forms of leukemia.
Based partly on this work, the UCSD-University of Pavia team sought to find out precisely how and where the enzyme complex bound to and interacted with SNAIL1, and why LSD1-CoREST is selectively drawn to, and recruited by, SNAIL1 in the first place.
Their research included analysis of the crystal structure of the LSD1-CoREST complex bound to SNAIL1 as determined from X-ray diffraction experiments. This snapshot demonstrated that the LSD1-CoRest complex tightly binds to a region of the SNAIL1 molecule that closely resembles the N-terminal tail of histone effectively mimicking the active site on this histone. The structure has been made publicly available in the Protein Data Bank.
“What this shows is how LSD1 recognizes and discriminates specific proteins in a crowded cell environment, and why the LSD1 complex is drawn to SNAIL 1,” said Andrea Mattevi, a researcher from the Department of Genetics and Microbiology at the University of Pavia, and the study’s principal investigator.
Though insightful, X-ray structures offer only a static view of molecular activity at a given moment. To learn more about the interaction of the enzyme complex and its target over time, scientists work with molecular dynamics software that simulate how proteins wiggle, weave and gyrate over time. Such is the complexity of the calculations needed for these simulations that researchers often turn to supercomputers.
“Experiments captured a key molecular-level photograph of this process from which computer simulations were initiated providing a movie on the nanosecond timescale,” added Baron. “For example, molecular films like these allow us to predict the routes of individual oxygen molecules to the reactive site of LSD1.”
Of particular note, the UCSD-University of Pavia researchers examined picosecond-by-picosecond movement of the LSD1-CoREST complex as it binds to SNAIL1, and changes – at the atomic level -- resulting from this activity. The “movies” show that oxygen can continue to reach the enzyme’s active site even when it’s bound to the histone tail, allowing the enzyme to perform its de-methylating task without needing to detach from its target.
“Overall, these observations and data are of crucial importance to understand which of these processes is the most promising target for future drugs,” said Mattevi. “Potent drugs could be developed targeting both the binding cleft of LSD1, as well as the active site access by oxygen molecules.”
Mattevi’s group in Pavia recently demonstrated that inhibitors to LSD1, and a close relative known as LSD2, strongly increased the potency of a chemotherapeutic agent called retinoic acid in the treatment of acute promyelocytic leukemia. Further, they recently discovered that known antidepressant drug inhibitors of enzymes with similar active sites (monoamine oxidase A and B) are promising candidates to develop highly specific inhibitors of LSD1 and LSD2.
Baron added he is in the process of establishing an independent research group focused on epigenetic drug discovery and design of LSD1 and LSD2 inhibitors using computational methodologies developed by the McCammon lab.
Provided by University of California - San Diego