A successful phonon calculation within the quantum Monte Carlo framework

The focus and ultimate goal of computational research in materials science and condensed matter physics is to solve the Schrödinger equation—the fundamental equation describing how electrons behave inside matter—exactly (without resorting to simplifying approximations). While experiments can certainly provide interesting insights into a material's properties, it is often computations that reveal the underlying physical mechanism. However, computations need not rely on experimental data and can, in fact, be performed independently, an approach known as "ab initio calculations." The density functional theory (DFT) is a popular example of such an approach.
For most material scientists and condensed matter physicists, DFT calculations are the bread and butter of their profession. However, despite being a powerful technique, DFT has had limited success with "strongly correlated materials"—materials with unusual electronic and magnetic properties. These materials, while interesting on their own, also possess technological useful properties, a fact that strongly motivates an ab initio framework suited to describe them.
To that end, a framework known as "ab initio quantum Monte Carlo" (QMC) has shown considerable promise and is expected to be the next generation of electronic structure calculations due to its superiority over DFT. However, even QMC is largely restricted to calculations of energy and atomic forces, limiting its utility in computing useful material properties.
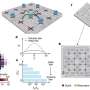
Now, in a breakthrough study published in Physical Review B (Editors' Suggestion), scientists have taken things to the next level based on an approach that allows them to reduce the statistical error in atomic force evaluation by two orders of magnitude and subsequently speeds up the computation by a factor of 104! "The drastic reduction in computational time will greatly expand the range of QMC calculations and enable highly accurate prediction of atomic properties of materials that have been difficult to handle," observes Assistant Professor Kousuke Nakano from Japan Advanced Institute of Science and Technology (JAIST), who, along with his colleagues Prof. Ryo Maezono from JAIST, Prof. Sandro Sorella from International School for advanced Studies (SISSA), Italy, and Dr. Tommaso Morresi and Prof. Michele Casula from Sorbonne Université, France, led this groundbreaking achievement.
The team applied their developed method to calculate the atomic vibrations of diamond, a typical reference material, as a proof-of-concept and showed that the results were consistent with experimental values. To perform these calculations, they used a large computer, Cray-XC40, located at the Research Center for Advanced Computing Infrastructure at Japan Advanced Institute of Science and Technology (JAIST), Japan, along with another located at RIKEN, Japan. The team made use of a QMC software package called "TurboRVB," initially launched by Prof. Sorella and Prof. Casula and developed later by Prof. Nakano along with others, to perform phonon dispersion calculations for diamond that were previously inaccessible, greatly expanding its scope.
Prof. Nakano looks forward to the applications of QMC in materials informatics (MI), a field dedicated to the design and search for novel materials using techniques of information science and computational physics. "While MI is currently governed by DFT, the rapid developments in computer performance, such as the exascale supercomputer, will help QMC gain popularity. In that regard, our developed method is going to be very useful for designing novel materials with real-life applications," concludes an optimistic Dr. Nakano.
More information: Kousuke Nakano et al, Atomic forces by quantum Monte Carlo: Application to phonon dispersion calculations, Physical Review B (2021). DOI: 10.1103/PhysRevB.103.L121110
Journal information: Physical Review B
Provided by Japan Advanced Institute of Science and Technology