December 4, 2020 report
NAD: Is nicotinamide adenine dinucleotide a super supplement or all hype?

NAD, or nicotinamide adenine dinucleotide, probably needs no introduction. Together with its primary alter-egos NADH, NADP and NADPH, our private suite of pyridine-based nucleotides serve as hydride donors in some 400 enzymatic reactions throughout the body. Beyond these signature dehydrogenase, hydroxylase and reductase reactions, other members of the larger NAD ecosystem function in receptor signaling pathways. Furthermore, the backbone NAD skeleton itself is extensively deployed in DNA repair, and directly consumed as additions to many other important molecules in different organelles.
Precursors and derivatives of NAD now adorn the shelves of pharmacies and supermarkets everywhere. These species include not just classical niacin (vitamin B3), but also other forms typically abbreviated as simply NA, NAM, NMN or NR. But what exactly are these molecules, and what, if anything, might they actually do for us?
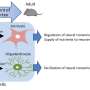
NAD concentrations throughout the body reflect a delicate balance between synthesis, consumption, transport and transformation. While NAD is created de novo from niacin using the so-called Preiss-Handler pathway in organs like the liver or kidney, many other tissues, such as as the nervous system, rely on salvage pathways using nicotinamide. Distribution of a proper bolus of NAD to the many far-flung synaptic locations of a neuron is ensured by motile mitochondria. While NAD has been studied for nearly 100 years, it was only this past September when the identity of the specific transporter that pumps NAD into mitochondria, SLC25A51, was formally uncovered.
NAD is consumed through the efforts of at least three different classes of enzymes: the poly ADP-ribose polymerases (PARPs), the NAD-dependent deacetylases (SIRTUINS), and NADases such as CD38. This latter molecule, CD38, is the subject of a flurry of papers recently published with considerable fanfare in Nature Metabolism. Taking a hint from previous findings that as the body ages, NAD markedly decreases and CD38 increases, these studies were collectively able to link these two processes together in a direct causal relationship.
The first paper, by Chini et al., demonstrates that CD38 expression in macrophages induced by senescence-associated inflammation is, in fact, the reason for age-related decline in NAD. From the abstract, the second paper, by Covarrubias et. al., appears to show much the same thing; however, the usually reliable Sci-Hub has been having some trouble loading that paper.
The canonical function of CD38 has traditionally been chalked up to generating a cyclic derivative of NAD known as cADPR. This function is vaguely reminiscent of the G-protein-receptor-driven cyclases which wrap up purine nucleotides into their cyclic forms. Additionally, when nicotinic acid is present under acidic conditions, CD38 can also hydrolyze NADP to NAADP. Depending on how you look at it, CD38 is either woefully inefficient at conserving NAD, or very efficient at liquidating it. It has been estimated that 100 molecules of NAD are required to generate just one molecule of cADPR. Inefficiency seems to be a rule in NAD systems, as researchers have determined that to make one gram of niacin from tryptophan, some 67 mg of the amino acid are required.
A curious feature of CD38 is its unexplained paradoxical membrane topology. In other words, most of the enzyme is configured in a type-II membrane orientation with its catalytic domain facing the extracellular compartment functioning as an ecto-NAD glycohydrolase. How, then, does it seemingly control intracellular NAD levels and calcium stores inside the cell? The answer, provided by Chini et. al., is that it short-circuits the entire works via extracellular degradation of the NAD precursor NMN.
In addition to type II CD38, there is also a type III version whose catalytic domain faces the cytosol. Cytosolic access is enabled either by having an upside-down orientation in the plasma membrane, or by persisting within the submembrane system of the cell. Type III CD38 is a nonglycosylated protein and differs from type II in that a disulfide bond is not formed in its carboxyl terminal residues. Antibodies like M19 can therefore be specifically made to recognize it. The protein itself is activated by crosslinking another set of cysteines found at positions 164 and 177. An NAD-family enzyme called NADPH-diaphorase 4 (Nox4) is responsible for this activation. It generates H2O2, which then facilitates the crosslink.
Is CD38 really the Boogeyman it has been made out to be?
CD38 has another, and in some ways more fascinating, cellular function: It moonlights as a master coordinator of mitochondrial transfer between cells. A few years ago, Stuart Rushworth and colleagues discovered that leukemic (AML) blast cells are able to survive and proliferate by coercing local bone marrow stem cells into feeding them functional mitochondria through tiny filaments known as tunneling nanotubes. AML cells do this by generating diffusible superoxide from yet another Nox enzyme, this time a Nox2. While blocking this transfer might seem like a potential way to treat cancers, inhibitors of Nox2 are not clinically available, and even if they were, blocking critical Nox signaling can be deadly in and of itself.
In the meantime, Hayakawa et. al. discovered that in vivo, astrocytes use a CD38-based mechanism to package mitochondria into vesicles for transfer to neurons. This process permits the neurons to survive tough times, particularly stroke. So perhaps CD38 isn't all bad. In other words, like cortisol, it might sometimes betray the body when called upon at too high a level in specific locations, but some baseline presence may be required for everyday normal function.
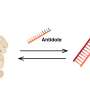
Mindful of these findings, Rushworth's group later found that elevated CD38 expression in his liquid tumor blood cells causes a similar mitochondrial transfer and rescue of these bad actors. Clinical trials for multiple myeloma are now underway to evaluate the merits of CD38 antibodies like isatuximab, which causes apoptosis directly, and daratumumab, which induces apoptosis indirectly. Newer antibodies from the Nature Metabolism articles above may also be of value here.
But what about neurons, do they have CD38? Do they donate mitochondria?
The surprising finding of the existence of another enzyme that produces cADPR and NAADP in neurons has not gone unnoticed. In this case, it wasn't CD38, but rather a molecule that has a completely different sequence. Known as SARM1, for "sterile α and armadillo motif–containing," it is a conserved member of the Toll-like receptor family and it appears to regulate axonal degeneration. Of particular interest, SARM1 has a unique localization sequence that targets it to mitochondria, where it has been associated with apoptosis.
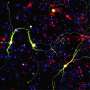
Until recently, it was thought that neurons only get rid of old or otherwise weak mitochondria. By ejecting spent mitochondria from their axons, apoptosis or other kinds of general malaise would presumably be avoided. This kind of outsourced mitophagy was notably discovered in retinal ganglion cells where the mitochondria were taken up by astrocytes and found to be degraded in LAMP1 (lysosomal associated protein) positive inclusions. However, more recent studies have shown that the altruistic donation of healthy mitochondria by neurons is really a thing, despite the many doubters and nay-sayers in the field. Using a co-culture system, Gao et. al. recently showed that astrocytes were able to increase the net membrane potential of their local mitochondrial stock by acquiring healthy reinforcements from nearby neurons. To do this, either CD38, or one of the two so-called MIRO proteins were required.
MIRO proteins adapt mitochondria to the motor proteins that transport them along cytoskeletal filaments. The authors also showed that this process breaks down in a condition known as Alexander disease (AxD). In AxD, astrocytes have a fault in their code for GFAP (glial fibrillary acidic protein), a member of the intermediate family of cytoskeletal proteins. The mitochondria that are transferred in normal individuals are not degraded in LAMP-positive lysosomes, and persist as high-functioning members within their new hosts' mitochondrial network. This represents the first case of a major disease whose cause can be directly traced to a deficiency in mitochondrial transfer.
Many studies have suggested that the ratios of NADH/NAD in the mitochondria and NADPH/NADP in the cytosol are the overarching status indicators for the cell at large. The addition of mitochondrial transfer into the mix offers many new ways to interpret how cells and organs adapt themselves to hold the requisite NAD-based tolerances. CD38 in particular represents both an attractive new therapeutic opportunity, and a unique insight into behind-the-scenes cooperation among cells.
More information: Claudia C. S. Chini et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD+ and NMN levels, Nature Metabolism (2020). DOI: 10.1038/s42255-020-00298-z
Anthony J. Covarrubias et al. Senescent cells promote tissue NAD+ decline during ageing via the activation of CD38+ macrophages, Nature Metabolism (2020). DOI: 10.1038/s42255-020-00305-3
Journal information: Nature Metabolism
© 2020 Science X Network