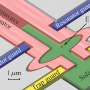
Quantum predictions
. Nickel ions (blue) and oxygen ions (red) surround a dopant ion of potassium (yellow). Quantum Monte Carlo methods can accurately predict regions where charge density (purple) will accumulate in these materials. Credit: Anouar Benali, Argonne National Laboratory")
Solving a complex problem quickly requires careful tradeoffs – and simulating the behavior of materials is no exception. To get answers that predict molecular workings feasibly, scientists must swap in mathematical approximations that speed computation at accuracy's expense.
But magnetism, electrical conductivity and other properties can be quite delicate, says Paul R.C. Kent of the Department of Energy's (DOE's) Oak Ridge National Laboratory. These properties depend on quantum mechanics, the movements and interactions of myriad electrons and atoms that form materials and determine their properties. Researchers who study such features must model large groups of atoms and molecules rather than just a few. This problem's complexity demands boosting computational tools' efficiency and accuracy.
That's where a method called quantum Monte Carlo (QMC) modeling comes in. Many other techniques approximate electrons' behavior as an overall average, for example, rather than considering them individually. QMC enables accounting for the individual behavior of all of the electrons without major approximations, reducing systematic errors in simulations and producing reliable results, Kent says.
Kent's interest in QMC dates back to his Ph.D. research at Cambridge University in the 1990s. At ORNL, he recently returned to the method because advances in both supercomputer hardware and in algorithms had allowed researchers to improve its accuracy.
"We can do new materials and a wider fraction of elements across the periodic table," Kent says. "More importantly, we can start to do some of the materials and properties where the more approximate methods that we use day to day are just unreliable."
Even with these advances, simulations of these types of materials, ones that include up to a few hundred atoms and thousands of electrons, requires computational heavy lifting. Kent leads a DOE Basic Energy Sciences Center, the Center for Predictive Simulations of Functional Materials (CPSFM) that includes researchers from ORNL, Argonne National Laboratory, Sandia National Laboratories, Lawrence Livermore National Laboratory, the University of California, Berkeley and North Carolina State University.
Their work is supported by a DOE Innovative and Novel Computational Impact on Theory and Experiments (INCITE) allocation of 140 million processor hours, split between Oak Ridge Leadership Computing Facility's Titan and Argonne Leadership Computing Facility's Mira supercomputers. Both computing centers are DOE Office of Science user facilities.
To take QMC to the next level, Kent and colleagues start with materials such as vanadium dioxide that display unusual electronic behavior. At cooler temperatures, this material insulates against the flow of electricity. But at just above room temperature, vanadium dioxide abruptly changes its structure and behavior.
Suddenly this material becomes metallic and conducts electricity efficiently. Scientists still don't understand exactly how and why this occurs. Factors such as mechanical strain, pressure or doping the materials with other elements also induce this rapid transition from insulator to conductor.
However, if scientists and engineers could control this behavior, these materials could be used as switches, sensors or, possibly, the basis for new electronic devices. "This big change in conductivity of a material is the type of thing we'd like to be able to predict reliably," Kent says.
Laboratory researchers also are studying these insulator-to-conductors with experiments. That validation effort lends confidence to the predictive power of their computational methods in a range of materials. The team has built open-source software, known as QMCPACK, that is now available online and on all of the DOE Office of Science computational facilities.
Kent and his colleagues hope to build up to high-temperature superconductors and other complex and mysterious materials. Although scientists know these materials' broad properties, Kent says, "we can't relate those to the actual structure and the elements in the materials yet. So that's a really grand challenge for the condensed-matter physics field."
The most accurate quantum mechanical modeling methods restrict scientists to examining just a few atoms or molecules. When scientists want to study larger systems, the computation costs rapidly become unwieldy. QMC offers a compromise: a calculation's size increases cubically relative to the number of electrons, a more manageable challenge. QMC incorporates only a few controlled approximations and can be applied to the numerous atoms and electrons needed. It's well suited for today's petascale supercomputers – capable of one quadrillion calculations or more each second – and tomorrow's exascale supercomputers, which will be at least a thousand times faster. The method maps simulation elements relatively easily onto the compute nodes in these systems.
The CPSFM team continues to optimize QMCPACK for ever-faster supercomputers, including OLCF's Summit, which will be fully operational in January 2019. The higher memory capacity on that machine's Nvidia Volta GPUs – 16 gigabytes per graphics processing unit compared with 6 gigabytes on Titan – already boosts computation speed. With the help of OLCF's Ed D'Azevedo and Andreas Tillack, the researchers have implemented improved algorithms that can double the speed of their larger calculations.
QMCPACK is part of DOE's Exascale Computing Project, and the team is already anticipating additional scaling challenges for running QMCPACK on future machines. To perform the desired simulations within roughly 12 hours on an exascale supercomputer, Kent estimates that they'll need algorithms that are 30 times more scalable than those within the current version.
Even with improved hardware and algorithms, QMC calculations will always be expensive. So Kent and his team would like to use QMCPACK to understand where cheaper methods go wrong so that they can improve them. Then they can save QMC calculations for the most challenging problems in materials science, Kent says. "Ideally we will learn what's causing these materials to be very tricky to model and then improve cheaper approaches so that we can do much wider scans of different materials."
The combination of improved QMC methods and a suite of computationally cheaper modeling approaches could lead the way to new materials and an understanding of their properties. Designing and testing new compounds in the laboratory is expensive, Kent says. Scientists could save valuable time and resources if they could first predict the behavior of novel materials in a simulation.
Plus, he notes, reliable computational methods could help scientists understand properties and processes that depend on individual atoms that are extremely difficult to observe using experiments. "That's a place where there's a lot of interest in going after the fundamental science, predicting new materials and enabling technological applications."
Provided by US Department of Energy