Scientists solve 3-D structure of cystic fibrosis protein in active, inactive states

Scientists at the UNC School of Medicine in collaboration with researchers at Oregon Health & Science University have resolved the three-dimensional molecular structure of the protein that's defective in people with cystic fibrosis in the protein's active and inactive state. The discovery, published in the journal Biochemistry, could open up new research avenues and help drug developers create enhanced pharmacotherapies to help people with CF.
Much of the biochemistry work was done in the lab of John Riordan, PhD, distinguished professor of biochemistry and biophysics at UNC-Chapel Hill. In the late 1980s, Riordan's lab discovered the mutated gene responsible for CF. If a child receives a copy of this faulty gene from each parent, the child will develop CF. The protein encoded by this gene was termed the cystic fibrosis transmembrane regulator, or CFTR, which is the chloride channel in epithelial cells that populate the respiratory system. People with CF lack a functional epithelial chloride channel, which is essential for maintaining the proper balance of salt and water in lungs and other organs. One result of this is the production of thick, sticky mucus that becomes difficult to move out of the airways, leading to chronic infections and a shorter lifespan for most people with CF.
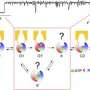
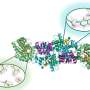
In the Riordan lab, postdoctoral fellow Jonathan Fay, PhD, led experiments using single-particle cryo-electron microscopy to discover the molecular structure of CFTR in the presence of ATP – a complex organic chemical necessary for many processes in cells, including a working chloride ion channel crucial for proper lung function. To help capture structures of the CFTR protein in its active and inactive state, the Riordan lab stabilized the CFTR protein such that the channel was off when dephosphorylated and locked-on when phosphorylated.

These molecular structures reveal a unique repositioning of parts of the CFTR protein, providing insights into the structural transition between active and inactive functional states of CFTR.
Moreover, Fay and colleagues observed details of this protein complex that differ from what other scientists have discovered in previous CFTR structures.
"It is really amazing how much cryo-EM technologies have advanced and how using these techniques can allow us to visualize different states of the channel," Fay said. "I think our results are very exciting. We discovered a new portal which presents a promising new area of the channel to target and control CFTR channel function."
And if researchers can successfully target and control the function of that channel, then they could create more precise therapies to better treat some people with CF.
More information: Jonathan F. Fay et al. Cryo-EM Visualization of an Active High Open Probability CFTR Anion Channel, Biochemistry (2018). DOI: 10.1021/acs.biochem.8b00763
Journal information: Biochemistry