June 21, 2017 report
Reconstructing chromosomal rearrangements of placental mammals over millions of years

(Phys.org)—A team of researchers from South Korea, the U.K. and the U.S. has used computational methods to follow chromosomal rearrangements in seven genomes. In their paper published in Proceedings of the National Academy of Sciences, the group describes the two-step process they followed that allowed them to better understand changes to chromosomal arrangement in certain mammals over time.
Scientists know that the way chromosomes are arranged changes over time as organisms evolve—but figuring out which changes occurred over millions of years for particular types of organisms has been difficult to see. In this new effort, the researchers undertook a two-step process that allowed them to see some of the ways that chromosomal arrangement changed in several genomes.
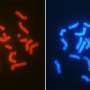
The research focused on the largest mammal type—placental mammals, which includes humans. The earliest evidence of such mammals dates back approximately 105 million years. To better understand how chromosomal arrangement has changed since that time, the researchers first conducted a comparative analysis of 19 genomes of several placental mammals. Doing so offered some clues regarding major changes that occurred in different species. To get an idea of the arrangement of the chromosomal state of the first placental mammals, team members wrote a computer program that reconstructed the chromosome based on the comparative analysis in the first step. The result was a report on 21 pairs of placental chromosomes.
The reconstructed chromosome produced by the software revealed that among other things, some chromosomal arrangements have stayed as they were over the course of 105 million years, while others changed dramatically. The researchers also saw instances of rearrangements in some sections and entire pieces swapped between other sections—they could actually see the breakpoints—162 of them in all.
The evidence also showed that chromosomal rearrangement was relatively slow in the first placental mammals, with just eight per 10 million years. The rate doubled at 65 million years for primates except for orangutans, which, the computer showed, have the closest chromosomal arrangement to our earliest ancestors.
In addition to offering new evidence of chromosomal changes over time, the research offers a path forward for studying other types of organisms and perhaps some clues regarding some modern diseases that involve chromosomal rearrangements.
More information: Reconstruction and evolutionary history of eutherian chromosomes, Jaebum Kim, PNAS, www.pnas.org/cgi/doi/10.1073/pnas.1702012114
Abstract
Whole-genome assemblies of 19 placental mammals and two outgroup species were used to reconstruct the order and orientation of syntenic fragments in chromosomes of the eutherian ancestor and six other descendant ancestors leading to human. For ancestral chromosome reconstructions, we developed an algorithm (DESCHRAMBLER) that probabilistically determines the adjacencies of syntenic fragments using chromosome-scale and fragmented genome assemblies. The reconstructed chromosomes of the eutherian, boreoeutherian, and euarchontoglires ancestor each included >80% of the entire length of the human genome, whereas reconstructed chromosomes of the most recent common ancestor of simians, catarrhini, great apes, and humans and chimpanzees included >90% of human genome sequence. These high-coverage reconstructions permitted reliable identification of chromosomal rearrangements over ∼105 My of eutherian evolution. Orangutan was found to have eight chromosomes that were completely conserved in homologous sequence order and orientation with the eutherian ancestor, the largest number for any species. Ruminant artiodactyls had the highest frequency of intrachromosomal rearrangements, and interchromosomal rearrangements dominated in murid rodents. A total of 162 chromosomal breakpoints in evolution of the eutherian ancestral genome to the human genome were identified; however, the rate of rearrangements was significantly lower (0.80/My) during the first ∼60 My of eutherian evolution, then increased to greater than 2.0/My along the five primate lineages studied. Our results significantly expand knowledge of eutherian genome evolution and will facilitate greater understanding of the role of chromosome rearrangements in adaptation, speciation, and the etiology of inherited and spontaneously occurring diseases.
Journal information: Proceedings of the National Academy of Sciences
© 2017 Phys.org