March 4, 2015 report
Color-coading gene sequences in human cells
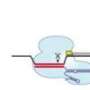
 ortholog-fluorescent proteins: S. pyogenes Sp dCas9, N. meningitidis Nm dCas9 and S. thermophilus St1 dCas9 fused to NLSs and under the control of the CMV-TetO promoter. The cognate sgRNAs Sp sgRNA, Nm sgRNA, and St1 sgRNA are under the control of the U6 promoter, and their sequences are shown below, with the mutations in red and hairpin extensions in orange. Directed by the appropriate sgRNA, different chromosomal loci are expected to become fluorescently labeled with any one of the three spectral versions of dCas9. In the diagram, such hypothetical targeted sequences are indicated by the color matching a particular dCas9/sgRNA ortholog. Credit: (c) 2015 PNAS, doi: 10.1073/pnas.1420024112")
(Phys.org)—Is there a way to peer inside the nucleus of a living cell and see how the genes interact? After the completion of the Human Genome Project in 2001, researchers have focused on epigenetic factors, spatial orientation, and non-coding repeated sequences, all of which affect genetic regulation and function. Hanhui Ma and Thoru Pederson from University of Massachusetts Medical School have demonstrated a new technique that resolves the spatial orientation between two target genetic repeated units within a living human cell. Their work is reported in the Proceedings of the National Academy of Sciences.
Ma and Pederson have done prior work labeling genes with a fluorescent protein tag using TALEs technology. However, the CRISPR/Cas9 system has proven to be the formidable approach to gene editing because it is able to target more than one genetic sequence at a time.
Other gene editing systems typically require a particular endonuclease to splice a specific gene, but for this system, Cas9 is used for all sequences, allowing for multiple targets within a single cell. Furthermore, to target a sequence, either for editing or labeling, researchers can make a synthetic RNA strand, known as the guide RNA, that is complementary to the target DNA sequence. While CRISPR/Cas9 has some limiting factors, such as off-target cuts or optimizing the PAM sequence, this is a robust method for targeting multiple genes.
In this experiment, Ma and Pederson used a catalytically inactive Cas9 (dCas9) endonculease to insert a fluorescent protein tag on the target sequence without causing a break in the DNA strand. Their goal was to insert two different labels that would target two different sequences. This would allow them to study the gene regions relative to each other as well as see where repeated locations occurred.
Their first step was to optimize their system. They chose to target the telomeric repeat for optimization and used three known Cas9 variants for the PAM sequences, S. pyogenes, Neisseria meningitidis, and Streptococcus thermophilus which were fused to red, green, and blue fluorescent proteins, respectively. Overall, the factors that they optimized were the choice of promotor driving the expression of dCas9, the number of fluorescent proteins fused in tandem, the length of guide RNAs, the PAM sequence choice, the choice of the sequence composition of the guide RNAs, and the number of nuclear localization signals. After optimizing these parameters, they were able to successfully observe all three targets.
They then tested whether two different CRISPR/Cas9 systems can simultaneously label the same sequence. They used S. pyogenes and Neisseria meningitidis systems and targeted one strand of the telomeric repeat. They identified two similar patterns showing two flourescence markers on the same genetic targets. They tested co-expression using S. pyogenes and Streptococcus thermophiles, and obtained similar results.
They then tested different, repeated chromosome-specific sequences. They chose two sequences on chromosome 9 and another sequence that is localized on chromosome 13. They were able to resolve the two sequences on chromosome 9 as being approximately two micrometers apart, which corresponds to the known distance between these sequences on the physical map of chromosome 9. They were also able to resolve two sequences between two different chromosomes, 9 and 13, that were even closer in proximity than the sequences on chromosome 9.
Finally, they wanted to test the resolution of their technique by choosing two sequences that are closer together on the same chromosome than their initial test with chromosome 9. They looked at two sequences on chromosome 13 that are known to be 1.9 Mbp apart from each other. They were able to resolve these using their multicolor CRISPR/Cas9 technique, and proved that it can resolve sequences that are 2.0 to 1.9 Mbp apart.
These experiments were able to resolve multiple interchromosome and intrachromosome target repeated sequences using flourescent protein tags. The authors are interested in looking at repeated sequence patterns that are specific for each chromosome. However, they envision this technique eventually being used for resolving two single-copy sequences.
More information: "Multicolor CRISPR labeling of chromosomal loci in human cells" Hanhui Ma, PNAS, DOI: 10.1073/pnas.1420024112
Abstract
The intranuclear location of genomic loci and the dynamics of these loci are important parameters for understanding the spatial and temporal regulation of gene expression. Recently it has proven possible to visualize endogenous genomic loci in live cells by the use of transcription activator-like effectors (TALEs), as well as modified versions of the bacterial immunity clustered regularly interspersed short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) system. Here we report the design of multicolor versions of CRISPR using catalytically inactive Cas9 endonuclease (dCas9) from three bacterial orthologs. Each pair of dCas9-fluorescent proteins and cognate single-guide RNAs (sgRNAs) efficiently labeled several target loci in live human cells. Using pairs of differently colored dCas9-sgRNAs, it was possible to determine the intranuclear distance between loci on different chromosomes. In addition, the fluorescence spatial resolution between two loci on the same chromosome could be determined and related to the linear distance between them on the chromosome's physical map, thereby permitting assessment of the DNA compaction of such regions in a live cell.
Journal information: Proceedings of the National Academy of Sciences
© 2015 Phys.org