Thousands of nuclear loci via target enrichment and genome skimming
The use of next-generation sequencing (NGS) technologies in phylogenetic studies is in a state of continual development and improvement. Though the botanically-inclined have historically focused on markers from the chloroplast genome, the importance of incorporating nuclear data is becoming increasingly evident. Nuclear genes provide not only the potential to resolve relationships between closely related taxa, but also the means to disentangle hybridization and better understand incongruences caused by incomplete lineage sorting and introgression.
By harnessing the power of NGS—which has increased sequencing capacity by several orders of magnitude over the past few years—scientists are now able to easily sequence enormous amounts of DNA or RNA from any genome within an organism, a practice that is transforming many areas of plant biology.
A team of international scientists, led by researchers at Oregon State University, has utilized a recently developed method to assemble a phylogenomic data set containing hundreds of nuclear loci and plastomes for milkweeds.
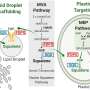
"This approach, termed Hyb-Seq, uses targeted sequence capture via hybridization-based enrichment and has shown great promise for obtaining large nuclear data sets," explains Dr. Aaron Liston, principal investigator of the study. "Sequencing low-copy nuclear genes has traditionally required a large amount of effort for each gene. Hyb-Seq eliminates the need for PCR optimization and cloning—two time-consuming and sometimes problematic steps."
The protocol is freely available in the September issue of Applications in Plant Sciences.
While it would be ideal to simply sequence entire genomes for every organism being studied, this is not yet feasible across large numbers of species. The Hyb-Seq approach reduces genomic complexity of the organism-of-interest by targeting only a small portion of the total genome. This is achieved by hybridizing DNA or RNA probes to specific regions of the genome, then simply discarding the remaining, unwanted regions.
"The probe design was done bioinformatically by comparing our draft sequence of the milkweed genome and transcriptome (expressed genes) to another genome in the same family and to genes that are conserved across the asterids and the angiosperms," explains Liston. "This allowed us to eliminate duplicated genes that can complicate phylogenetic inference and select relatively conserved genes, so that they could be obtained from divergent milkweed species with a single probe set."
This approach enabled Liston and colleagues to sequence over 700 genes for 10 Asclepias species and two related genera. "Furthermore," says Liston, "we were able to assemble complete plastomes from the off-target reads."
"It is likely that as sequencing technology advances, it will be feasible in the next decade or so to sequence complete genomes routinely and inexpensively. However, until that time, the ability to sequence hundreds of genes at a time—as is possible with the Hyb-Seq method—represents a significant and exciting advance over previous methods."
More information: Weitemier, K., S. C. K. Straub, R. C. Cronn, M. Fishbein, R. Schmickl, A. McDonnell, and A. Liston. 2014. Hyb-Seq: Combining target enrichment and genome skimming for plant phylogenomics. Applications in Plant Sciences 2(9): 1400042. DOI: 10.3732/apps.1400042
Journal information: Applications in Plant Sciences
Provided by American Journal of Botany