January 29, 2014 report
Light field microscopy for whole brain activity maps

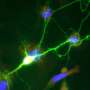
(Phys.org) —Advances in light-sheet microscopy have led to impressive images and videos of the brain in action. With this technique, a plane of light is scanned through the sample to excite fluorescent calcium sensors which proxy neural activity. While the transparent head of a baby zebrafish, or even the whole body of a tiny worm can now be mapped with impressive lateral resolution, the temporal resolution is still limited by the need for the scanning. A potentially much more powerful technique—light field microscopy (LFM)—gets rid of the moving parts and can therefore create volumetric images in a fraction of the time. Ed Boyden and his collaborators have now tuned these methods to create the first "functional LFM images" of the brain. Their results have just been published to the Arxiv preprint server.
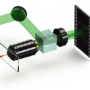
One of the early pioneers of LFM was Marc Levoy, who many folks may have heard of due to his groundbreaking work at Google. His many projects include co-designing their book scanner, launching Street View, and contributions Google glass. Together with his collaborators, Marc also refined the techniques that led to the imaging system used in the popular Lytro camera, which like LFM can be refocussed after-the-fact to different parts of an image. To create the LFM they placed a microlens array beyond the specimen at the intermediate image plane of a conventional microscope. Different points on the sample are then recorded in separate small images on a light field plane further above the sample. Deconvolution techniques can then used to build up whole images.
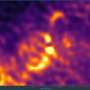
While the LFM has been used before for in-vitro imaging, Ed's new work represents the first time that spatial resolutions comparable to the groundbreaking zebrafish brain activity maps (courtesy of Misha Ahrens) have been presented. I asked Ed how his new work in imaging the entire c. elegans worm compares with the light sheet methods used by Ahrens and company. He stressed that eliminating the physical component, (the actual scan) is the key to the superb temporal resolution now possible, and that the deconvolution algorithms yield the improved spatial resolution.
While the zebrafish results were acquired in a volume 800x600x200 microns per side, with an impressive lateral resolution of 0.65 um, the refresh rate was a relatively slow 1.3 seconds. With Boyden's LFM, the imaging volume used was slightly smaller (~350 μm x 350 μm x 30 μm) although that is fine here because basically, the entire nervous system of the worm, in fact most of its body, could fit in that volume. The resolution was a respectable ~1.4μm in the lateral dimension and 2.6μm in the axial, still good enough for single neuron precision. The impressive thing here though is the whole volume image rate—up to 50 hz. That is fast enough to shift the temporal resolution problem away from the hardware, and put it back in the laps of the chemists that make the dyes or protein sensors used for imaging.
At the end of the day these mapping techniques are really only as useful as the questions one can ask, and answer, using them. Last year's fascinating images of a zebrafish watching its lunch swim by as a cerebral doppelganger of fish activity percolated eerily through its own brain was proof enough of this. Ed and his group were already able to image the complete set of head ganglia neurons in the worm at the same time selected ventral cord control units were imaged, not a bad start. Not having any real experience myself, as far as how these techniques are actually implemented into the experimental workflow, I asked neurophysiologist David Schoppik to comment here. In his words:
"This work from the Vaziri and Boyden laboratories is both important and exciting, and is a nice complement to ongoing work in the Levoy lab (Broxton et. al., 2013). While a light-field microscope can acquire data breathtakingly quickly, processing times of 20-90 minutes/frame (i.e. weeks of processing to analyze a couple minutes of acquired data) limits its applicability. As hardware and algorithms mature, this is sure to improve. Perhaps most excitingly, as the next generation of genetically-encoded voltage sensors comes online, light-field microscopy unique holds the promise of acquisition of large volumes with the speed necessary to capture membrane potential dynamics."
To David's point, we are already beginning to see some of these next generation voltage sensors take shape. Intriguing work on multi-state "Flash Memory" sensors that can be toggled on and off by different wavelengths of light have just been published. Some of the fancier techniques, like direct genetic encoding, enable the sensor to be targeted to just a small number of specific cells. This is crucial so that the total number of signal producers in the field of view is not so large that everything is bright. At the same time however, as we have learned using techniques like Brainbow II, if you only have labelled synapses, you don't have the whole picture. Glia, blood vessels, and organelles are all critical pieces of the puzzle.
However now, we are beginning to understand that much of the larger activity of the nervous system, particularly at the periphery, is not something we can readily comprehend in isolation from the immediate tissue in which it embeds. Something as simple as tactile sensing in the worm, whether by raw nerve ending or whatever version of a vibratory corpuscular sensor a worm might possess, are really larger chemo-mechanical issues that extend beyond nerve membranes. Imaging the entire body of the worm, down to the level of movements of cell organelles should give a fuller picture of integrated activity beyond transient calcium or voltage spikes.
If functions like adaptation and memory, as many now believe, involve structurally observable change—actual motions by and within cells—then perhaps these slower dynamics will come to be appreciated on par with the faster spikes associated more with direct signalling. The previously unimagined abilities of cells (including neurons) to target and exchange mitochondria, for example, might be imaged in worms and correlated directly to spiking activity.
It may be a slight digression from microscopy techniques, but if we consider an extreme case of organelle transport, we might have some guidance for what to look for in new experiments. The 10-meter long motor axons of the blue whale, for example, need to provide fresh organelles that offer proteins and power to the end effectors where they are needed. However doing this on any kind of timescale relevant to spikes or synaptic modifications is not just difficult, it is impossible. Slow phase axonal transport would take decades to reach their muscle target points, while even the fast component pool (perhaps several mm/day) would still take months. Furthermore, consider here that during the growth phase, the whale axons tethered to the fluke region are being pulled aft at the astounding rate of 3cm per DAY. That imposes a huge "volume debt" that must be satisfied by some source of cellular materials.
The point of this diversion is that if in larger creatures, neurons are materially isolated from their targets on the timescales of electrical activity, than local tissue interactions, like sharing mitochondria and other packages of genetic information, most be significant. For now, the humble worm, together with these new LFM methods offers an ideal platform to further probe these kinds of activities from the molecular to tissue level. As they give way to in vivo imaging of larger brains, nerves, and bodies, we should have a clearer picture of what we might count as "activity" in the full functional map of a calculating organism.
More information: Simultaneous whole-animal 3D-imaging of neuronal activity using light field microscopy, arXiv:1401.5333 [physics.optics] arxiv.org/abs/1401.5333
Abstract
3D functional imaging of neuronal activity in entire organisms at single cell level and physiologically relevant time scales faces major obstacles due to trade-offs between the size of the imaged volumes, and spatial and temporal resolution. Here, using light-field microscopy in combination with 3D deconvolution, we demonstrate intrinsically simultaneous volumetric functional imaging of neuronal population activity at single neuron resolution for an entire organism, the nematode Caenorhabditis elegans. The simplicity of our technique and possibility of the integration into epi-fluoresence microscopes makes it an attractive tool for high-speed volumetric calcium imaging.
Journal information: arXiv
© 2014 Phys.org