Sequencing without PCR reduces bias in measuring biodiversity
DNA barcode sequencing without the amplification of DNA by PCR beats the problem of false positives which can inflate estimates of biodiversity, finds a study published in BioMed Central and BGI Shenzhen's open access journal GigaScience. This method tested on a bulk 'squashome' of mixed insect samples is also able rapidly and cost-effectively estimate biomass.
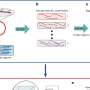
Often samples collected in the field are too small be sequenced directly. Traditionally, to get round this problem, DNA from the sample is amplified using multiple cycles of PCR. However this process is highly dependent on the chemical and physical properties of each piece of DNA and consequently this approach can selectively amplify certain sequences more than others.
Researchers from BGI and the China National GeneBank-Shenzhen have developed a method of COI metabarcoding which uses mitochondrial enrichment prior to DNA extraction, rather than PCR, to ensure a high enough concentration of DNA for next generation ultra-deep sequencing to be accurate.
Successfully tested on 69 arthropods collected from a mountain in sub-tropical China this method was able to correctly match insects to their reference samples without introducing the false positives often seen using traditional methods. False positives are the main culprit behind biodiversity inflation. A few samples from insects with a body length of less than 5mm contained too little material for this method to work.
The screen also managed to identify bacteria within the samples, two of which were Wolbachia, a symbiont frequently found in insects, plus Legionellaceae and Bartonellaceae which can cause disease in humans. Additionally DNA was found from a member of the Lepidoptera order (butterflies and moths) even though it was not in the original insect mixture. It is thought that this probably either came from the gut of an insect that ate it, or from an undetected fragment or egg.
Commenting on the ability of this study in evaluating biodiversity Dr Xin Zhou who led this study commented, "We also found that sequencing 'volume' (nucleotide numbers) strongly indicated total biomass for the species in a bulk sample. By being able to identify both species and the biomass for each species more accurately than traditional PCR-based sequencing, this method could revolutionise biodiversity research and biomonitoring."
More information: Ultra-deep sequencing enables high-fidelity recovery of biodiversity for bulk arthropod samples without PCR amplification, Xin Zhou, Yiyuan Li, Shanlin Liu, Qing Yang, Xu Su, Lili Zhou, Min Tang, Ribei Fu, Jiguang Li and Quanfei Huang GigaScience 2013, 2:4 doi:10.1186/2047-217X-2-4
NGS biodiversity data
Zhou, X; Li, Y; Liu, S; Yang, Q; Su, X; Zhou, L; Tang, M; Fu, R; Li, J GigaScience Database 2013 dx.doi.org/10.5524/100045
NGS Biodiversity software
Zhou, X; Li, Y; Liu, S; Yang, Q; Su, X; Zhou, L; Tang, M; Fu, R; Li, J; Huang, Q, GigaScience Database 2013 dx.doi.org/10.5524/100046 (Provides software and supporting material)
Journal information: GigaScience
Provided by BioMed Central